Homework Week 7
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CHAPTER 12: Thermodynamics Why Chemical Reactions Happen
4/3/2015 CHAPTER 12: Thermodynamics Why Chemical Reactions Happen A tiny fraction of the sun's energy is used to produce complicated, ordered, high- Useful energy is being "degraded" in energy systems such as life the form of unusable heat, light, etc. • Our observation is that natural processes proceed from ordered, high-energy systems to disordered, lower energy states. • In addition, once the energy has been "degraded", it is no longer available to perform useful work. • It may not appear to be so locally (earth), but globally it is true (sun, universe as a whole). 1 4/3/2015 Thermodynamics - quantitative description of the factors that drive chemical reactions, i.e. temperature, enthalpy, entropy, free energy. Answers questions such as- . will two or more substances react when they are mixed under specified conditions? . if a reaction occurs, what energy changes are associated with it? . to what extent does a reaction occur to? Thermodynamics does NOT tell us the RATE of a reaction Chapter Outline . 12.1 Spontaneous Processes . 12.2 Entropy . 12.3 Absolute Entropy and Molecular Structure . 12.4 Applications of the Second Law . 12.5 Calculating Entropy Changes . 12.6 Free Energy . 12.7 Temperature and Spontaneity . 12.8 Coupled Reactions 4 2 4/3/2015 Spontaneous Processes A spontaneous process is one that is capable of proceeding in a given direction without an external driving force • A waterfall runs downhill • A lump of sugar dissolves in a cup of coffee • At 1 atm, water freezes below 0 0C and ice melts above 0 0C • Heat flows -

Entropy and Free Energy
Spontaneous Change: Entropy and Free Energy 2nd and 3rd Laws of Thermodynamics Problem Set: Chapter 20 questions 29, 33, 39, 41, 43, 45, 49, 51, 60, 63, 68, 75 The second law of thermodynamics looks mathematically simple but it has so many subtle and complex implications that it makes most chemistry majors sweat a lot before (and after) they graduate. Fortunately its practical, down-to-earth applications are easy and crystal clear. We can build on those to get to very sophisticated conclusions about the behavior of material substances and objects in our lives. Frank L. Lambert Experimental Observations that Led to the Formulation of the 2nd Law 1) It is impossible by a cycle process to take heat from the hot system and convert it into work without at the same time transferring some heat to cold surroundings. In the other words, the efficiency of an engine cannot be 100%. (Lord Kelvin) 2) It is impossible to transfer heat from a cold system to a hot surroundings without converting a certain amount of work into additional heat of surroundings. In the other words, a refrigerator releases more heat to surroundings than it takes from the system. (Clausius) Note 1: Even though the need to describe an engine and a refrigerator resulted in formulating the 2nd Law of Thermodynamics, this law is universal (similarly to the 1st Law) and applicable to all processes. Note 2: To use the Laws of Thermodynamics we need to understand what the system and surroundings are. Universe = System + Surroundings universe Matter Surroundings (Huge) Work System Matter, Heat, Work Heat 1. -

Atkins' Physical Chemistry
Statistical thermodynamics 2: 17 applications In this chapter we apply the concepts of statistical thermodynamics to the calculation of Fundamental relations chemically significant quantities. First, we establish the relations between thermodynamic 17.1 functions and partition functions. Next, we show that the molecular partition function can be The thermodynamic functions factorized into contributions from each mode of motion and establish the formulas for the 17.2 The molecular partition partition functions for translational, rotational, and vibrational modes of motion and the con- function tribution of electronic excitation. These contributions can be calculated from spectroscopic data. Finally, we turn to specific applications, which include the mean energies of modes of Using statistical motion, the heat capacities of substances, and residual entropies. In the final section, we thermodynamics see how to calculate the equilibrium constant of a reaction and through that calculation 17.3 Mean energies understand some of the molecular features that determine the magnitudes of equilibrium constants and their variation with temperature. 17.4 Heat capacities 17.5 Equations of state 17.6 Molecular interactions in A partition function is the bridge between thermodynamics, spectroscopy, and liquids quantum mechanics. Once it is known, a partition function can be used to calculate thermodynamic functions, heat capacities, entropies, and equilibrium constants. It 17.7 Residual entropies also sheds light on the significance of these properties. 17.8 Equilibrium constants Checklist of key ideas Fundamental relations Further reading Discussion questions In this section we see how to obtain any thermodynamic function once we know the Exercises partition function. Then we see how to calculate the molecular partition function, and Problems through that the thermodynamic functions, from spectroscopic data. -

Review Entropy: a Measure of the Extent to Which Energy Is Dispersed
Review entropy: a measure of the extent to which energy is dispersed throughout a system; a quantitative (numerical) measure of disorder at the nanoscale; given the symbol S. In brief, processes that increase entropy (ΔS > 0) create more disorder and are favored, while processes that decrease entropy (ΔS < 0) create more order and are not favored. Examples: 1. Things fall apart over time. (constant energy input is required to maintain things) 2. Water spills from a glass, but spilled water does not move back into the glass. 3. Thermal energy disperses from hot objects to cold objects, not from cold to hot. 4. A gas will expand to fill its container, not concentrate itself in one part of the container. Chemistry 103 Spring 2011 Energy disperses (spreads out) over a larger number of particles. Ex: exothermic reaction, hot object losing thermal energy to cold object. Energy disperses over a larger space (volume) by particles moving to occupy more space. Ex: water spilling, gas expanding. Consider gas, liquid, and solid, Fig. 17.2, p. 618. 2 Chemistry 103 Spring 2011 Example: Predict whether the entropy increases, decreases, or stays about the same for the process: 2 CO2(g) 2 CO(g) + O2(g). Practice: Predict whether ΔS > 0, ΔS < 0, or ΔS ≈ 0 for: NaCl(s) NaCl(aq) Guidelines on pp. 617-618 summarize some important factors when considering entropy. 3 Chemistry 103 Spring 2011 Measuring and calculating entropy At absolute zero 0 K (-273.15 °C), all substances have zero entropy (S = 0). At 0 K, no motion occurs, and no energy dispersal occurs. -

Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions
CHEM 1B: GENERAL CHEMISTRY Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions Instructor: Dr. Orlando E. Raola 20-1 Santa Rosa Junior College Chapter 20 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20-2 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20.1 The Second Law of Thermodynamics: Predicting Spontaneous Change 20.2 Calculating Entropy Change of a Reaction 20.3 Entropy, Free Energy, and Work 20.4 Free Energy, Equilibrium, and Reaction Direction 20-3 Spontaneous Change A spontaneous change is one that occurs without a continuous input of energy from outside the system. All chemical processes require energy (activation energy) to take place, but once a spontaneous process has begun, no further input of energy is needed. A nonspontaneous change occurs only if the surroundings continuously supply energy to the system. If a change is spontaneous in one direction, it will be nonspontaneous in the reverse direction. 20-4 The First Law of Thermodynamics Does Not Predict Spontaneous Change Energy is conserved. It is neither created nor destroyed, but is transferred in the form of heat and/or work. DE = q + w The total energy of the universe is constant: DEsys = -DEsurr or DEsys + DEsurr = DEuniv = 0 The law of conservation of energy applies to all changes, and does not allow us to predict the direction of a spontaneous change. 20-5 DH Does Not Predict Spontaneous Change A spontaneous change may be exothermic or endothermic. Spontaneous exothermic processes include: • freezing and condensation at low temperatures, • combustion reactions, • oxidation of iron and other metals. -

AP ® 2007–2008 Professional Development Workshop Materials
AP ® Chemistry 2007–2008 Professional Development Workshop Materials Special Focus: Thermochemistry The College Board: Connecting Students to College Success Th e College Board is a not-for-profi t membership association whose mission is to connect students to college success and opportunity. Founded in 1900, the association is composed of more than 5,000 schools, colleges, universities, and other educational organizations. Each year, the College Board serves seven million students and their parents, 23,000 high schools, and 3,500 colleges through major programs and services in college admissions, guidance, assessment, fi nancial aid, enrollment, and teaching and learning. Among its best-known programs are the SAT®, the PSAT/ NMSQT®, and the Advanced Placement Program® (AP®). Th e College Board is committed to the principles of excellence and equity, and that commitment is embodied in all of its programs, services, activities, and concerns. For further information, visit www.collegeboard.com. Pages 7, 9 and 10: Silberberg, Martin. Chemistry: Th e Molecular Nature of Matter and Change. 4th ed. New York: McGraw-Hill, 2006. pg. 226-228. Reprinted with Permission by Th e McGraw-Hill Companies, Inc. Page 9: Chang, Raymond and Brandon Cruickshank. Chemistry. 8th ed. New York: McGraw-Hill, 2005. Reprinted with Permission by Th e McGraw-Hill Companies, Inc. Page 23: Hess’s Law Lab reprinted from Tim Allen’s Computer Science/Chemistry Homepage, http://www.geocities.com/tjachem/mgo.html Th e College Board acknowledges and wishes to thank all contributors for permission to reprint the copyrighted material identifi ed in the text. Sources not included in the captions or body of the text are listed here. -

Heat Capacity and Thermodynamic Properties of Equilibrium Sulfur to the Temperature 388.36 K, and the Heat Capacity of Calorimetry Conference Coppera
UNITED STATES DEPARTMENT OF THE INTERIOR GEOLOGICAL SURVEY Heat capacity and thermodynamic properties of equilibrium sulfur to the temperature 388.36 K, and the heat capacity of Calorimetry Conference coppera by Bruce S. Hemingway U.S. Geological Survey Open-File Report 99-324 a This report is preliminary and has not been reviewed for conformity with U.S. Geological Survey editorial standards Abstract The heat capacity of specially prepared orthorhombic sulfur has been measured in a low-temperature adiabatic calorimeter. Measurements from T ~ 6 K to near the melting temperature transition at Tfug = 388.36 K are reported for equilibrium sulfur: for the orthorhombic modification from T » 6 K to the temperature of the orthorhombic-to-monoclinic transition Ttrg = 368.3 K, and for the monoclinic modification from Ttrs to Tfus . The molar entropy AjS^/R and molar enthalpy function A^.H^/RT for orthorhombic sulfur calculated from this data set are (3.843 ± 0.010) and (1.776 ± 0.005), respectively, where T = 298.15 K, T 1 - 0, and R = 8.31451 J-K^-mol' 1 . Four measurements of the, enthalpy of the orthorhombic to monoclinic phase transition were made with three samples. At Ttrs , the average value for the enthalpy of transition AcrsH° is (401.3 ± 0.8 J-mol" 1 ) . The heat capacity of the orthorhombic phase is given by the equation: Cp<n/ (J-K^-mol-1 ) = 15.830 + 0.023036 (T/K) for the temperature interval (290 to 368.3) K, and that of the monoclinic phase by Cp, m/ (J-K^-mol- 1 ) = 11.8498 + 0.035197 (T/K) for the temperature interval (368.3 to 388.36) K. -

4 Standard Entropies of Hydration of Ions
View Article Online / Journal Homepage / Table of Contents for this issue 4 Standard Entropies of Hydration of Ions By Y. MARCUS and A. LOEWENSCHUSS Department of Inorganic and Analytical Chemistry, The Hebrew University of Jerusalem, 9 I904 Jerusalem, Israel 1 Introduction Since ions are hydrated in aqueous solutions, the standard thermodynamic functions of the hydration process are of interest. Of these, the standard entropy of hydration is expected to shed some light on the state of the ion and the surrounding aqueous medium. In particular, the notion of water- structure-breaking and -making is amenable to quantitative expression in terms of a structural entropy that can be derived from the experimental standard molar entropy of hydration. The process of hydration of an ion X', where z is the algebraic charge number of the ion, consists of its transfer from the ideal gas phase to the aqueous phase at infinite dilution: X'(g) - X'(aq) (1) A thorough discussion of the general process of solvation, of which hydration Published on 01 January 1984. Downloaded by FAC DE QUIMICA 27/07/2015 16:16:07. is a particular case, was recently published by Ben-Naim and Marcus,' with a sequel on the solvation of dissociating electrolytes by Ben-Naim.2 Provided that the number density concentration scale or an equivalent one (e.g., the molar, i.e., mol dm-') is employed, the standard molar entropy change of the process, conventionally determined experimentally and converted appro- priately to an absolute value, equals Avogadro's number NA, times the entropy change per particle.'-2The conventional standard states are the ideal gas at 0.1 MPa pressure (formerly at 0.101 325 MPa = 1 atm pressure) for the gas phase (g) and the ideal aqueous solution under 0.1 MPa pressure and at 1 mol dmP3concentration of the ion for the aqueous phase. -

Thermodynamics and Chemistry
THERMODYNAMICS AND CHEMISTRY SECOND EDITION HOWARD DEVOE Thermodynamics and Chemistry Second Edition Version 10, January 2020 Howard DeVoe Associate Professor of Chemistry Emeritus University of Maryland, College Park, Maryland [email protected] The first edition of this book was previously published by Pearson Education, Inc. It was copyright ©2001 by Prentice-Hall, Inc. The second edition, version 10 is copyright ©2020 by Howard DeVoe. This work is licensed under a Creative Commons Attribution 4.0 International License: https://creativecommons.org/licenses/by/4.0/ The book was typeset using the LATEX typesetting system and the memoir class. Most of the figures were produced with PSTricks, a related software program. The fonts are Adobe Times, MathTime, and Computer Modern Typewriter. A Solutions Manual is available at the Web site linked below. I thank the Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland for hosting the Web site for this book: http://www.chem.umd.edu/thermobook SHORT CONTENTS Biographical Sketches 15 Preface to the Second Edition 16 From the Preface to the First Edition 17 1 Introduction 19 2 Systems and Their Properties 27 3 The First Law 57 4 The Second Law 105 5 Thermodynamic Potentials 138 6 The Third Law and Cryogenics 153 7 Pure Substances in Single Phases 166 8 Phase Transitions and Equilibria of Pure Substances 195 9 Mixtures 225 10 Electrolyte Solutions 287 11 Reactions and Other Chemical Processes 304 12 Equilibrium Conditions in Multicomponent Systems 367 13 The Phase Rule -

Thermodynamic Investigations of Some Aqueous Solutions
THERMODYNAMIC INVESTIGATIONS OF SOME AQUEOUS SOLUTIONS THROUGH CALORIMETRY AND DENSIMETRY ROBERT A. MARRIOTT B.Sc., University of Lethbridge, 1998 A Thesis Submitted to the Council on Graduate Studies of the University of Lethbridge in Partial Fulfilment of the Requirements for the Degree MASTER OF SCIENCE LETHBRIDGE, ALBERTA July, 1999 ©Robert A. Marriott, 1999 FOR GRAMPA BOLIVAR iii ABSTRACT Relative densities and heat capacity ratios have been measured for selected aqueous systems. These measurements have been used to calculate apparent molar volumes and heat capacities. Densities of aqueous sodium bromide have been measured from 374 to 522 K and 10.00 to 30.00 MPa using a recently developed high temperature and pressure vibrating tube densimeter. These data have been used to test the utility of an automated high temperature and pressure densimetric data analysis program. Apparent molar volumes and heat capacities of several aqueous rare earth sulphate systems at 298.15 K and 0.10 MPa have been reported, and discussed in terms of ionic contributions. Single ion partial molar volumes and heat capacities for aqueous trivalent rare earth species have been estimated in a review of apparent molar data from the literature and through the use of a semi-empirical Debye-Hiickel equation. These single ion properties have subsequently been used to estimate the single ion properties of the monosulphate and disulphate rare earth complex species. Rigorous relaxation calculations are presented in a discussion of apparent molar heat capacities, where relaxation contributions are shown to be negative. Apparent molar volumes and densities for aqueous L-histidine, L-phenylalanine, L-tyrosine, L-tryptophan, and L-dopa have been used to estimate reported partial molar properties at infinite dilution and at 288.15,298.15,313.15, and 328.15 K. -
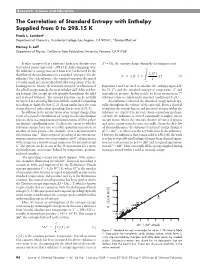
The Correlation of Standard Entropy with Enthalpy Supplied from 0 to 298.15 K
Research: Science and Education The Correlation of Standard Entropy with Enthalpy Supplied from 0 to 298.15 K Frank L. Lambert* Department of Chemistry, Occidental College, Los Angeles, CA 90041; *[email protected] Harvey S. Leff Department of Physics, California State Polytechnic University, Pomona, CA 91768 It takes energy to heat a substance from near absolute zero T → 0 K, the entropy change during the heating process is to standard room temperature, 298.15 K. As heating progresses, the substance’s entropy increases from zero (as dictated by the T p third law of thermodynamics) to a standard entropy for the Cp T S° Sp z S T p dT (2) substance. For each substance, the standard entropy is the sum of T reversible small increments dH divided by temperature T for the 0 heating process. That is, the standard entropyS° is a function of Equations 1 and 2 are used to calculate the enthalpy input ΔH° the added energy, namely, the total enthalpy ∆H° delivered dur- for (0, T°) and the standard entropy at temperature T° and ing heating. This energy spreads spatially throughout the solid atmospheric pressure. In this article, we focus attention on 77 and is stored within it. The entropy function can be usefully substances that are solids under standard conditions (5–23). interpreted as a spreading function, with the symbol S connoting As a substance is heated, the absorbed energy spreads spa- spreading, as clarified below(1, 2). Along similar lines, the term tially throughout the volume of the material. From a classical energy dispersal, rather than spreading, has been used (3, 4). -

Spontaneous Processes
The first law: transformation of energy into heat and work Chemical reactions can be used to provide heat and for doing work. Compare fuel value of different compounds. What drives these reactions to proceed in the direction they do? Why does combustion proceed spontaneously? Why does NaCl dissolve spontaneously in water? Spontaneous Processes A spontaneous process is one that occurs by itself, given enough time, without external intervention Spontaneous for T > 0oC Spontaneous for T < 0oC Reversible vs Irreversible Processes Reversible processes Are at equilibrium Driving force is only infinitesimally greater than the opposing force Process occurs in a series of infinitesimal steps, and at each step the system in at equilibrium with the surroundings Would take an infinite amount of time to carry out Irreversible Process Not at equilibrium; a spontaneous process Reversal cannot be achieved by changing some variable by an infinitesimal amount Which direction will an irreversible process proceed to establish equilibrium? What thermodynamic properties determine the direction of spontaneity? The change in enthalpy during a reaction is an important factor in determining whether a reaction is favored in the forward or reverse direction. Are exothermic reactions more likely to be spontaneous than endothermic? Not necessarily - for example, melting of ice is spontaneous above 0oC and is endothermic. Entropy Consider the following expansion of a gas into a vacuum. When the valve is open, there are four possible arrangements or STATES; all states are equal in energy Opening the valve allows this system of two particles, more arrangements; higher degree of DISORDER. As the number of particles increases in the system, the number of possible arrangements that the system can be in increases Processes in which the disorder of the system increases tend to occur spontaneously.