Dimenhydrinate)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
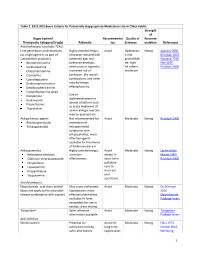
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Antiemetics/Antivertigo Agents
Antiemetic Agents Therapeutic Class Review (TCR) May 1, 2019 No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected]. May 2019 Proprietary Information. Restricted Access – Do not disseminate or copy without approval. © 2004-2019 Magellan Rx Management. All Rights Reserved. 3 FDA-APPROVED INDICATIONS Drug Manufacturer Indication(s) NK1 receptor antagonists aprepitant capsules generic, Merck In combination with other antiemetic agents for: (Emend®)1 . -

Scopolamine As a Potential Treatment Option in Major Depressive Disorder - a Literature Review
Isr J Psychiatry - Vol. 58 - No 1 (2021) Scopolamine as a Potential Treatment Option in Major Depressive Disorder - A Literature Review Dusan Kolar, MD, MSc, PhD, FRCPC Department of Psychiatry, Queen’s University, Mood Disorders Research and Treatment Service, Kingston, Ontario, Canada as selective serotonin reuptake inhibitors (SSRIs) and ABSTRACT serotonin norepinephrine reuptake inhibitors (SNRIs), are currently used as first-line pharmacological treat- Introduction: Slow onset response to antidepressants ments for major depressive disorder (MDD). Despite the and partial response is a common problem in mood availability of a comprehensive list of antidepressants, disorders psychiatry. There is an ongoing need for rapid- approximately 50% of the patients on an antidepressant acting antidepressants, particularly after introducing regimen experience non-response to treatment (3, 4). ketamine in the treatment of depression. Scopolamine Furthermore, the mood elevating effects of antidepres- may have promise as an antidepressant. sant medication is often delayed (5). It is recommended Methods: The author conducted a literature review to that the patient be treated for at least six weeks with identify available treatment trials of scopolamine in the adequate dosage of the given antidepressant before unipolar and bipolar depression in PubMed, the Cochrane considering making changes to the treatment (5). Partial database, Ovid Medline and Google Scholar. to no response to treatment with antidepressants and delayed onset of action indicate that there is a need for Results: There have been eight treatment trials of the development of novel and improved medications scopolamine in MDD and bipolar depression. Seven for the treatment of depression. Many studies in recent studies confirmed significant antidepressant effects years have recognised two different classes of drugs with of scopolamine used as monotherapy and as an rapid and robust antidepressant effects. -

(Antimuscarinic) Drugs?
© July - August 2018 How well do you know your anticholinergic (antimuscarinic) drugs? nticholinergic drugs, prescribed for a variety of clini- Acal conditions, are amongst the most frequently used prescription drugs in BC (Table 1). Also referred to as “an- timuscarinics,” such drugs specifically block muscarinic receptors for acetylcholine (ACh).1 Muscarinic ACh recep- tors are important in the parasympathetic nervous system that governs heart rate, exocrine glands, smooth muscles, clude drugs whose active metabolites are potent- as well as brain function. In contrast, nicotinic ACh recep- ly antimuscarinic,5 or which often cause typical tors stimulate contraction of striated muscles. This Letter is AC adverse effects such as dry mouth or urinary intended to remind clinicians of commonly used drugs that retention.6 People taking antihistamines, antide- have anticholinergic (AC), or technically, antimuscarinic pressants, antipsychotics, opioids, antimuscarinic properties, and of their potential adverse effects. inhalers, or many other drugs need to know that Beneficial and harmful effects of anticholinergic drugs have blockade of ACh receptors can cause bothersome been known for centuries. In Homer’s Odyssey, the nymph or even dangerous adverse effects (Table 3). pharmacologist Circe utilized central effects of atropinics Subtle and not-so-subtle toxicity in the common plant jimson weed (Datura stramonium) to cause delusions in the crew of Odysseus. Believing they Students often learn the adverse effects of anticho- had been turned into pigs, they could be herded.2 linergics from a mnemonic, e.g.: “Blind as a bat, Sometimes a drug is recommended specifically for its an- mad as a hatter, red as a beet, hot as a hare, dry as ticholinergic potency. -

The Promise of Ketamine
Neuropsychopharmacology (2015) 40, 257–258 & 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 www.neuropsychopharmacology.org Editorial Circumspectives: The Promise of Ketamine William A Carlezon*,1 and Tony P George2 1 2 Department of Psychiatry, Harvard Medical School, McLean Hospital, Belmont, MA, USA; Department of Psychiatry, University of Toronto, Toronto, Ontario, Canada Neuropsychopharmacology (2015) 40, 257–258; doi:10.1038/npp.2014.270 In this issue we reprise a Feature called Circumspectives. offering hope that fast-acting but safe antidepressants are The general format of a Circumspectives article is similar to possible. a debate, with separate sections in which two thought Despite growing enthusiasm for ketamine and its leaders articulate their individual positions on a topic of promise, Dr Schatzberg describes some sobering details great importance to our community of researchers. The and gaps in knowledge. Ketamine is a drug of abuse and, distinguishing element, however, is that the piece ends with despite some exceptionally elegant studies on the mechan- a ‘reconciliation’ that is co-authored by both and includes ism (eg, Li et al, 2010; Autry et al, 2011), there is no ideas or experiments that will move the field forward. consensus on how it produces therapeutic effects. As one The current Circumspectives (Sanacora and Schatzberg, example, Dr Schatzberg points out similarities in some of 2015) is entitled ‘Ketamine: Promising Path or False the molecular actions of ketamine and scopolamine, Prophecy in the Development of Novel Therapeutics for another familiar and long-standing member of our phama- Mood Disorders?’. It is co-authored by Gerard Sanacora and copea shown to produce rapid antidepressant effects (Furey Alan F Schatzberg, who are leaders in this field. -

Drugs That Can Cause Delirium (Anticholinergic / Toxic Metabolites)
Drugs that can Cause Delirium (anticholinergic / toxic metabolites) Deliriants (drugs causing delirium) Prescription drugs . Central acting agents – Sedative hypnotics (e.g., benzodiazepines) – Anticonvulsants (e.g., barbiturates) – Antiparkinsonian agents (e.g., benztropine, trihexyphenidyl) . Analgesics – Narcotics (NB. meperidine*) – Non-steroidal anti-inflammatory drugs* . Antihistamines (first generation, e.g., hydroxyzine) . Gastrointestinal agents – Antispasmodics – H2-blockers* . Antinauseants – Scopolamine – Dimenhydrinate . Antibiotics – Fluoroquinolones* . Psychotropic medications – Tricyclic antidepressants – Lithium* . Cardiac medications – Antiarrhythmics – Digitalis* – Antihypertensives (b-blockers, methyldopa) . Miscellaneous – Skeletal muscle relaxants – Steroids Over the counter medications and complementary/alternative medications . Antihistamines (NB. first generation) – diphenhydramine, chlorpheniramine). Antinauseants – dimenhydrinate, scopolamine . Liquid medications containing alcohol . Mandrake . Henbane . Jimson weed . Atropa belladonna extract * Requires adjustment in renal impairment. From: K Alagiakrishnan, C A Wiens. (2004). An approach to drug induced delirium in the elderly. Postgrad Med J, 80, 388–393. Delirium in the Older Person: A Medical Emergency. Island Health www.viha.ca/mhas/resources/delirium/ Drugs that can cause delirium. Reviewed: 8-2014 Some commonly used medications with moderate to high anticholinergic properties and alternative suggestions Type of medication Alternatives with less deliriogenic -

Stability and Compatibility of Combinations of Hydromorphone
The Canadian Journal of Hospital Pharmacy - Volume 46, No. 2, April 1993 61 Stability and Compatibility of Combinations oman of Hydromorphone and Dimenhydrinate, ations Lorazepam or Prochlorperazine Jnally >hoto- Scott E. Walker, John Iazzetta, Carlo De Angelis and Danny W.C. Lau laser :ii, in figure ABSTRACT RESUME .Each The stability and compatibility of combinations of hydro On a melange diverses solutions d'hydromorphone .rabic morphone (2, 10 and 40 mg/ml) admixed separately with (2, 10 et 40 mg/ml) avec, separemen( un volume egal :onse- dimenhydrinate (50 mg/ml), prochlorperazine (5 mg/ml), d'une solution soit de dimenhydrate (50 mglmL), de 1ld be or lorazepam (4 mg/ml) were tested over a seven-day prochlorperazine (5 mglmL) ou de lorazepam (4 mglmL), Jarate period at 4°C, 23°C and 37°C In addition to visual pour verifier la compatibilite de ces trois medicaments avec lished inspection and pH, the concentration of each component l'hydromorphone et determiner la stabilite du melange ie ac in the binary mixture was determined by a stability binaire pendant sept jours a 4°C, a 23°C et a 37°C ion to indi.cating lifjuid chromatographic method Each test was En plus d'effectuer wz examen visuel et de deter ipt. completed at time zero, one, four, six and seven days after miner le pH, on a dose !es composants des melanges par mixing equal volumes of each medication. une methode de chromatographie en phase lifjuide indi The hydromorphone-dimenhydrinate combination was quant la stabilite. On a analyse !es melanges au moment compatible and stable for 24 hours. -

The Anticholinergic Toxidrome
Poison HOTLINE Partnership between Iowa Health System and University of Iowa Hospitals and Clinics July 2011 The Anticholinergic Toxidrome A toxidrome is a group of symptoms associated with poisoning by a particular class of agents. One example is the opiate toxidrome, the triad of CNS depression, respiratory depression, and pinpoint pupils, and which usually responds to naloxone. The anticholinergic toxidrome is most frequently associated with overdoses of diphenhydramine, a very common OTC medication. However, many drugs and plants can produce the anticholinergic toxidrome. A partial list includes: tricyclic antidepressants (amitriptyline), older antihistamines (chlorpheniramine), Did you know …… phenothiazines (promethazine) and plants containing the anticholinergic alkaloids atropine, hyoscyamine and scopolamine (Jimson Weed). Each summer, the ISPCC receives approximately 10-20 The mnemonic used to help remember the symptoms and signs of this snake bite calls, some being toxidrome are derived from the Alice in Wonderland story: from poisonous snakes (both Blind as a Bat (mydriasis and inability to focus on near objects) local and exotic). Red as a Beet (flushed skin color) Four poisonous snakes can be Hot as Hades (elevated temperature) found in Iowa: the prairie These patients can sometimes die of agitation-induced hyperthermia. rattlesnake, the massasauga, Dry as a Bone (dry mouth and dry skin) the copperhead, and the Mad as a Hatter (hallucinations and delirium) timber rattlesnake. Each Bowel and bladder lose their tone (urinary retention and constipation) snake has specific territories Heart races on alone (tachycardia) within the state. ISPCC A patient who has ingested only an anticholinergic substance and is not specialists have access to tachycardic argues against a serious anticholinergic overdose. -

Ce4less.Com Ce4less.Com Ce4less.Com Ce4less.Com Ce4less.Com Ce4less.Com Ce4less.Com
Hallucinogens And Dissociative Drug Use And Addiction Introduction Hallucinogens are a diverse group of drugs that cause alterations in perception, thought, or mood. This heterogeneous group has compounds with different chemical structures, different mechanisms of action, and different adverse effects. Despite their description, most hallucinogens do not consistently cause hallucinations. The drugs are more likely to cause changes in mood or in thought than actual hallucinations. Hallucinogenic substances that form naturally have been used worldwide for millennia to induce altered states for religious or spiritual purposes. While these practices still exist, the more common use of hallucinogens today involves the recreational use of synthetic hallucinogens. Hallucinogen And Dissociative Drug Toxicity Hallucinogens comprise a collection of compounds that are used to induce hallucinations or alterations of consciousness. Hallucinogens are drugs that cause alteration of visual, auditory, or tactile perceptions; they are also referred to as a class of drugs that cause alteration of thought and emotion. Hallucinogens disrupt a person’s ability to think and communicate effectively. Hallucinations are defined as false sensations that have no basis in reality: The sensory experience is not actually there. The term “hallucinogen” is slightly misleading because hallucinogens do not consistently cause hallucinations. 1 ce4less.com ce4less.com ce4less.com ce4less.com ce4less.com ce4less.com ce4less.com How hallucinogens cause alterations in a person’s sensory experience is not entirely understood. Hallucinogens work, at least in part, by disrupting communication between neurotransmitter systems throughout the body including those that regulate sleep, hunger, sexual behavior and muscle control. Patients under the influence of hallucinogens may show a wide range of unusual and often sudden, volatile behaviors with the potential to rapidly fluctuate from a relaxed, euphoric state to one of extreme agitation and aggression. -

Hyoscyamine, Atropine, Scopolamine, and Phenobarbital
PATIENT & CAREGIVER EDUCATION Hyoscyamine, Atropine, Scopolamine, and Phenobarbital This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Donnatal; Phenohytro What is this drug used for? It is used to treat irritable bowel syndrome. It is used to treat ulcer disease. It may be given to your child for other reasons. Talk with the doctor. What do I need to tell the doctor BEFORE my child takes this drug? If your child is allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell the doctor about the allergy and what signs your child had. If your child has any of these health problems: Heart problems due to bleeding, bowel block, enlarged colon, glaucoma, a hernia Hyoscyamine, Atropine, Scopolamine, and Phenobarbital 1/9 that involves your stomach (hiatal hernia), myasthenia gravis, slow-moving GI (gastrointestinal) tract, trouble passing urine, or ulcerative colitis. If your child has ever had porphyria. If your child has been restless or overexcited in the past after taking phenobarbital. If your child is addicted to drugs or alcohol, or has been in the past. This is not a list of all drugs or health problems that interact with this drug. Tell the doctor and pharmacist about all of your child’s drugs (prescription or OTC, natural products, vitamins) and health problems. You must check to make sure that it is safe to give this drug with all of your child’s other drugs and health problems. -

Chapter 34 • Drugs Used to Treat Nausea and Vomiting
• Chapter 34 • Drugs Used to Treat Nausea and Vomiting • Learning Objectives • Compare the purposes of using antiemetic products • State the therapeutic classes of antiemetics • Discuss scheduling of antiemetics for maximum benefit • Nausea and Vomiting • Nausea : the sensation of abdominal discomfort that is intermittently accompanied by a desire to vomit • Vomiting (emesis): the forceful expulsion of gastric contents up the esophagus and out of the mouth • Regurgitation : the rising of gastric or esophageal contents to the pharynx as a result of stomach pressure • Common Causes of Nausea and Vomiting • Postoperative nausea and vomiting • Motion sickness • Pregnancy Hyperemesis gravidarum: a condition in pregnancy in which starvation, dehydration, and acidosis are superimposed on the vomiting syndrome • Common Causes of Nausea and Vomiting (cont’d) • Psychogenic vomiting: self-induced or involuntary vomiting in response to threatening or distasteful situations • Chemotherapy-induced emesis (CIE) Anticipatory nausea and vomiting: triggered by sight and smell associated with treatment Acute CIE: stimulated directly by chemotherapy 1 to 6 hours after treatment Delayed emesis: occurs 24 to 120 hours after treatment; may be induced by metabolic by-products of chemotherapy • Drug Therapy for Selected Causes of Nausea and Vomiting • Postoperative nausea and vomiting (PONV) • Antiemetics include: Dopamine antagonists Anticholinergic agents Serotonin antagonists H2 antagonists (cimetidine, ranitidine) • Nursing Process for Nausea and Vomiting -

Motion Sickness Traveler Summary Key Points Motion Sickness Consists of a Group of Signs and Symptoms That Develop in Response to Real Or Perceived Motion
Motion Sickness Traveler Summary Key Points Motion sickness consists of a group of signs and symptoms that develop in response to real or perceived motion. Symptoms include dizziness, nausea, and vomiting. Prevention includes: Eating light meals Avoiding alcohol Sitting in the front seat of a car, over the wings on an airplane, or mid-deck on ships and facing forward in buses and trains Avoiding tasks requiring a close focus (e.g., reading) Using over-the-counter (OTC) or prescription medication OTC medications include: Dimenhydrinate, diphenhydramine, cyclizine, or meclizine: take 1 hour before departure and continue during the trip. These medications can cause sedation; do not mix with alcohol. Read labels carefully. Check for cautions regarding use in certain conditions. Prescription medications include: Scopolamine patches: place behind the ear; change every 3 days; apply 8 hours before the first incidence of rough weather or rough roads. Dry mouth and dry eyes may result. Patches do not work if cut in half. More than 1 patch should never be applied; hallucinations or psychosis may result. Strong sedatives (such as promethazine or prochlorperazine): take orally or by suppository after onset of severe symptoms, but anticipate sleep for a number of hours. A cruise medical clinic may administer injectable promethazine if absolutely necessary. Introduction The human body has a delicate system of equilibrium that relies on fluids in the inner ear, visual sensors, and other physical input to maintain a sense of balance. When incoming signals are in conflict—for example, when the body is at rest yet the eyes sense movement—this system is disturbed, causing the symptoms of motion sickness.