Identi Cation of Novel Hub Genes Associated with Gastric Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Profiling of Cell Cycle Genes of Breast Cells Exposed to Etodolac
1383-1391.qxd 29/3/2010 11:48 Ì ™ÂÏ›‰·1383 ONCOLOGY REPORTS 23: 1383-1391, 2010 Profiling of cell cycle genes of breast cells exposed to etodolac D. ROY1,2*, G.A. ARASON2*, B. CHOWDHURY3,4, A. MITRA5 and G.M. CALAF6,7 1Department of Natural Sciences, Hostos College of the City University of New York, Bronx, NY; 2Graduate Biotechnology Program, Manhattan College, Riverdale, NY; 3Human Retrovirus Section, Vaccine Branch, National Cancer Institute, National Institutes of Health, Frederick, MD; 4Department of Cellular Injury, Military Casualty Research, Walter Reed Army Institute of Research, Silver Spring, MD; 5Department of Chemistry, The College of Mount Saint Vincent, Riverdale, NY; 6Center for Radiological Research, Columbia University Medical Center, New York, NY, USA; 7Instituto de Alta Investigación, Universidad de Tarapaca, Arica, Chile Received September 29, 2009; Accepted November 6, 2009 DOI: 10.3892/or_00000775 Abstract. Breast cancer represents the second leading cause Introduction of cancer-related deaths in the world. There is increasing evidence that perturbation of cell cycle regulation is an Breast cancer represents the second leading cause of cancer- important contributing factor to various cancer progression related deaths in the United States and other Western countries; stages. There are key checkpoints in the cell cycle involving accounting for about 30-40% of all newly diagnosed cancers various regulatory proteins. The relationship between these (1,2). Hereditary breast cancer accounts for just 5-10% of cell cycle regulatory proteins and cell cycle arrest by cyclo- cases. It is generally believed that a family history of breast oxygenase (COX) inhibitors during neoplastic progression cancer and hormonal imbalances also contributes to the remains largely unknown. -

The Mitotic Checkpoint Protein MAD2 Delivers Monoamine Transporters to Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.09.447721; this version posted June 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The mitotic checkpoint protein MAD2 delivers monoamine transporters to endocytosis. Florian Koban1*, Michael Freissmuth1 https://orcid.org/0000-0003-0912-3114, https://orcid.org/0000-0001-9398-1765 1Institute of Pharmacology and the Gaston H. Glock Research Laboratories for Exploratory Drug Development, Center of Physiology and Pharmacology, Medical University of Vienna, Vienna, Austria. *Corresponding Author: Florian Koban, PhD Institute of Pharmacology and the Gaston H. Glock Research Laboratories for Exploratory Drug Development, Center of Physiology and Pharmacology, Medical University of Vienna. Waehringer Strasse 13A, 1090 Vienna, Austria E-mail: [email protected] Tel.: +43 1 40160 31328 Author contributions: F. K. conceptualized the study; F. K. conducted research; F. K. and M. F. analyzed data; F.K. and M.F. wrote the paper. bioRxiv preprint doi: https://doi.org/10.1101/2021.06.09.447721; this version posted June 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 2 ABSTRACT Monoamine transporters retrieve serotonin (SERT), dopamine (DAT) and norepinephrine (NET) from the synaptic cleft. Surface levels of transporters are also regulated by internalization. Clathrin-mediated endocytosis of cargo proteins requires adaptor protein 2 (AP2), which recruits cargo to the nascent clathrin- cage. The transporter C-terminus is required for internalization but lacks an AP2- binding site. -

Gene Section Review
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Review MAD2L1 (mitotic arrest deficient 2, yeast, human homolog like-1) Elizabeth M. Petty, Kenute Myrie Division of Medical Genetics Departments of Human Genetics and Internal Medicine University of Michigan Medical School 1150 West Medical Center Drive, 4301 MSRB III, Ann Arbor, Michigan 48109- 0638, USA (EMP, KM) Published in Atlas Database: March 2001 Online updated version : http://AtlasGeneticsOncology.org/Genes/MAD2L1ID304.html DOI: 10.4267/2042/37729 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2001 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Other names: HsMAD2; MAD2; MAD2A HGNC (Hugo): MAD2L1 Shadded boxes (1-5) depict the 5 exons of MAD2L1. The black Location : 4q27 triangle indicates a del A mutation that was found in the CAL51 Local order: As noted on the GM99-GB4 breast cancer cell line. Open triangkes depict the locations of Chromosome 4 map: Position: 548.24 (cR3000) Lod identified sequence variants. Figure is not drawn to scale. score: 1.16 Reference Interval: D4S2945-D4S430 Transcription (115.1-125.1 cM) It is located within the NCBI BAC MAD2L1 has 5 coding exons. No alternative splicing genomic contig: NT_006302.2 which is part of the has been described. Regulation of its transcription in homo sapiens chromosome 4 sequence segment. human cells is currently poorly understood. Note: MAD2L1 was intially (and errounously) mapped by fluorescence in situ hybridization (FISH) to 5q23- Protein q31. Subsequent comprehensive mapping studies using somatic cell hybrid analysis, radiation hybrid (RH) Description mapping, and FISH localized it to 4q27. -

Protein Interactions in the Cancer Proteome† Cite This: Mol
Molecular BioSystems View Article Online PAPER View Journal | View Issue Small-molecule binding sites to explore protein– protein interactions in the cancer proteome† Cite this: Mol. BioSyst., 2016, 12,3067 David Xu,ab Shadia I. Jalal,c George W. Sledge Jr.d and Samy O. Meroueh*aef The Cancer Genome Atlas (TCGA) offers an unprecedented opportunity to identify small-molecule binding sites on proteins with overexpressed mRNA levels that correlate with poor survival. Here, we analyze RNA-seq and clinical data for 10 tumor types to identify genes that are both overexpressed and correlate with patient survival. Protein products of these genes were scanned for binding sites that possess shape and physicochemical properties that can accommodate small-molecule probes or therapeutic agents (druggable). These binding sites were classified as enzyme active sites (ENZ), protein–protein interaction sites (PPI), or other sites whose function is unknown (OTH). Interestingly, the overwhelming majority of binding sites were classified as OTH. We find that ENZ, PPI, and OTH binding sites often occurred on the same structure suggesting that many of these OTH cavities can be used for allosteric modulation of Creative Commons Attribution 3.0 Unported Licence. enzyme activity or protein–protein interactions with small molecules. We discovered several ENZ (PYCR1, QPRT,andHSPA6)andPPI(CASC5, ZBTB32,andCSAD) binding sites on proteins that have been seldom explored in cancer. We also found proteins that have been extensively studied in cancer that have not been previously explored with small molecules that harbor ENZ (PKMYT1, STEAP3,andNNMT) and PPI (HNF4A, MEF2B,andCBX2) binding sites. All binding sites were classified by the signaling pathways to Received 29th March 2016, which the protein that harbors them belongs using KEGG. -

Proteomic Analysis of Ubiquitin Ligase KEAP1 Reveals Associated Proteins That Inhibit NRF2 Ubiquitination
Published OnlineFirst February 4, 2013; DOI: 10.1158/0008-5472.CAN-12-4400 Cancer Molecular and Cellular Pathobiology Research Proteomic Analysis of Ubiquitin Ligase KEAP1 Reveals Associated Proteins That Inhibit NRF2 Ubiquitination Bridgid E. Hast1, Dennis Goldfarb2, Kathleen M. Mulvaney1, Michael A. Hast4, Priscila F. Siesser1, Feng Yan1, D. Neil Hayes3, and Michael B. Major1,2 Abstract Somatic mutations in the KEAP1 ubiquitin ligase or its substrate NRF2 (NFE2L2) commonly occur in human cancer, resulting in constitutive NRF2-mediated transcription of cytoprotective genes. However, many tumors display high NRF2 activity in the absence of mutation, supporting the hypothesis that alternative mechanisms of pathway activation exist. Previously, we and others discovered that via a competitive binding mechanism, the proteins WTX (AMER1), PALB2, and SQSTM1 bind KEAP1 to activate NRF2. Proteomic analysis of the KEAP1 protein interaction network revealed a significant enrichment of associated proteins containing an ETGE amino acid motif, which matches the KEAP1 interaction motif found in NRF2. Like WTX, PALB2, and SQSTM1, we found that the dipeptidyl peptidase 3 (DPP3) protein binds KEAP1 via an "ETGE" motif to displace NRF2, thus inhibiting NRF2 ubiquitination and driving NRF2-dependent transcription. Comparing the spectrum of KEAP1-interacting proteins with the genomic profile of 178 squamous cell lung carcinomas characterized by The Cancer Genome Atlas revealed amplification and mRNA overexpression of the DPP3 gene in tumors with high NRF2 activity but lacking NRF2 stabilizing mutations. We further show that tumor-derived mutations in KEAP1 are hypomorphic with respect to NRF2 inhibition and that DPP3 overexpression in the presence of these mutants further promotes NRF2 activation. -

Expression and Mutational Analyses of the Human MAD2L1 Gene in Breast Cancer Cells
GENES,CHROMOSOMES&CANCER29:356–362(2000) BRIEFCOMMUNICATION ExpressionandMutationalAnalysesoftheHuman MAD2L1GeneinBreastCancerCells MelanieJ.Percy,1 KenuteA.Myrie,2 ChristopherK.Neeley,1 JamesN.Azim,1 StephenP.Ethier,3 and ElizabethM.Petty1,2* 1DepartmentofInternalMedicine,UniversityofMichiganMedicalCenter,AnnArbor,Michigan 2DepartmentofHumanGenetics,UniversityofMichiganMedicalCenter,AnnArbor,Michigan 3DepartmentofRadiationOncology,UniversityofMichiganComprehensiveCancerCenter,AnnArbor,Michigan Breastcancerisaheterogeneousdisorderinwhichmosttumorsdisplaysomedegreeofaneuploidy,especiallythoseatlater stagesofthedisease.Aneuploidyandassociatedchromosomeinstabilitymaybeimportantintheprogressionofmammary tumorigenesis.Aneuploidyispreventedduringnormalcelldivisioninpartthroughregulationofamitoticspindlecheckpoint wheremitoticarrestpreventssegregationofmisalignedchromosomesintodaughtercellsatanaphase.Mitoticarrestgenes, includingtheMADfamily,whichwasoriginallycharacterizedinyeast,helpregulatenormalfunctionofthemitoticspindle checkpoint.DecreasedexpressionofthehumangeneMAD2L1waspreviouslyreportedinabreastcancercelllineexhibiting chromosomeinstabilityandaneuploidy.ToexplorefurtherthepotentialroleofMAD2L1inbreastcancer,weanalyzed MAD2L1geneexpressionin13minimallytogrosslyaneuploidhumanbreastcancercelllinesandfoundsignificantdifferences ofexpressioninthreelines.SequenceanalysisofMAD2L1cDNAintheseaswellasnineadditionalaneuploidbreastcancer andfiveimmortalizednormalhumanmammaryepithelialcelllinesrevealedoneheterozygousframeshift(572delA)mutation inacancercelllinethatdemonstratedahighleveloftranscriptexpression.Inaddition,two3ЈUTRsequencevariantswere -

MAD1 (MAD1L1) (NM 003550) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC203471L4V MAD1 (MAD1L1) (NM_003550) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: MAD1 (MAD1L1) (NM_003550) Human Tagged ORF Clone Lentiviral Particle Symbol: MAD1L1 Synonyms: MAD1; PIG9; TP53I9; TXBP181 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) ACCN: NM_003550 ORF Size: 2154 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC203471). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_003550.2 RefSeq Size: 2754 bp RefSeq ORF: 2157 bp Locus ID: 8379 UniProt ID: Q9Y6D9 Protein Families: Druggable Genome Protein Pathways: Cell cycle MW: 83.1 kDa This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 MAD1 (MAD1L1) (NM_003550) Human Tagged ORF Clone Lentiviral Particle – RC203471L4V Gene Summary: MAD1L1 is a component of the mitotic spindle-assembly checkpoint that prevents the onset of anaphase until all chromosome are properly aligned at the metaphase plate. -
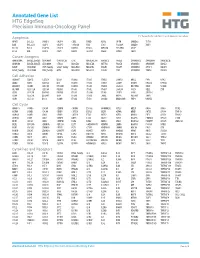
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -
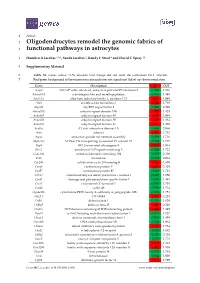
Oligodendrocytes Remodel the Genomic Fabrics of Functional Pathways in Astrocytes
1 Article 2 Oligodendrocytes remodel the genomic fabrics of 3 functional pathways in astrocytes 4 Dumitru A Iacobas 1,2,*, Sanda Iacobas 3, Randy F Stout 4 and David C Spray 2,5 5 Supplementary Material 6 Table S1. Genes whose >1.5x absolute fold-change did not meet the individual CUT criterion. 7 Red/green background of the expression ratio indicates not significant (false) up-/down-regulation. Gene Description X CUT Acap2 ArfGAP with coiled-coil, ankyrin repeat and PH domains 2 -1.540 1.816 Adamts18 a disintegrin-like and metallopeptidase -1.514 1.594 Akr1c12 aldo-keto reductase family 1, member C12 1.866 1.994 Alx3 aristaless-like homeobox 3 1.536 1.769 Alyref2 Aly/REF export factor 2 -1.880 2.208 Ankrd33b ankyrin repeat domain 33B 1.593 1.829 Ankrd45 ankyrin repeat domain 45 1.514 1.984 Ankrd50 ankyrin repeat domain 50 1.628 1.832 Ankrd61 ankyrin repeat domain 61 1.645 1.802 Arid1a AT rich interactive domain 1A -1.668 2.066 Artn artemin 1.524 1.732 Aspm abnormal spindle microtubule assembly -1.693 1.716 Atp6v1e1 ATPase, H+ transporting, lysosomal V1 subunit E1 -1.679 1.777 Bag4 BCL2-associated athanogene 4 1.723 1.914 Birc3 baculoviral IAP repeat-containing 3 -1.588 1.722 Ccdc104 coiled-coil domain containing 104 -1.819 2.130 Ccl2 chemokine -1.699 2.034 Cdc20b cell division cycle 20 homolog B 1.512 1.605 Cenpf centromere protein F 2.041 2.128 Cep97 centrosomal protein 97 -1.641 1.723 COX1 mitochondrially encoded cytochrome c oxidase I -1.607 1.650 Cpsf7 cleavage and polyadenylation specific factor 7 -1.635 1.891 Crct1 cysteine-rich -

Increased Expression of the Genes for Mitotic Spindle Assembly and Chromosome Segregation in Both Lung and Pancreatic Carcinomas
CANCER GENOMICS & PROTEOMICS 1: 231-240 (2004) Increased Expression of the Genes for Mitotic Spindle Assembly and Chromosome Segregation in Both Lung and Pancreatic Carcinomas KENJI HAMADA1, MICHIO UEDA3, MASAMI SATOH2, NAOHITO INAGAKI1, HIROSHI SHIMADA3 and HISAFUMI YAMADA-OKABE1 1Pharmaceutical Research Department IV, Kamakura Research Laboratories, Chugai Pharmaceutical Co. Ltd., 200 Kajiwara, Kamakura, Kanagawa 247-8530; 2Department of Surgery, Tohoku University, 4-1 Seiryoucho, Aobaku, Sendai, Miyagi 980-8575; 3Department of Surgery, School of Medicine, Yokohama City University, 3-9 Fukuura, Kanazawa, Yokohama, Kanagawa 236-0004, Japan Abstract. Genes whose expression was modulated in two feature of at least lung carcinomas and pancreatic carcinomas different tumor types, lung or pancreatic carcinoma, were and, therefore, such genes may be potential targets for widely identified by DNA microarray and subsequent expression effective anticancer agents. correlation analyses. For more accurate comparison of the gene expression between tumor and normal cells, tumor cells Lung cancer and pancreatic cancer are among the most and normal epithelium cells were isolated by laser-captured frequent causes of cancer death worldwide. Although new microdissection. Genes whose expression was significantly molecular-targeting drugs such as Iressa and Gemzar have altered in lung carcinomas or pancreatic carcinomas as been introduced into their treatment, the efficacy of compared to their normal counterparts were ranked by the T- chemotherapeutic agents against these cancers is still values calculated from the Fisher’s ratios and their limited (1, 2). Since lung and pancreatic cancers are often corresponding background Fisher’s ratios, followed by asymptomatic in their early stages, most patients have statistical confirmation using the Welch’s t-test. -

Usbiological Datasheet
MAD2L1 (Mitotic spindle assembly Checkpoint Protein MAD2A, HsMAD2, Mitotic arrest deficient 2-like Protein 1, MAD2-like Protein 1, MAD2) Catalog number 223989 Supplier United States Biological Cell cycle progression is subject to arrest at the mitotic spindle assembly checkpoint in response to incorrect spindle fiber assembly. MAD2 (for mitotic arrest-deficient) is a component of the mitotic spindle checkpoint. Cells with mutated MAD2 do not undergo mitotic arrest in response to incorrect spindle fiber assembly, which results in missegregation and eventual cell death. A breast carcinoma cell line with reduced MAD2 expression, T47D, was shown to complete mitosis in the presence of nocodazole, an inhibitor of mitotic spindle assembly. MAD2 is localized to unattached kinetochores during prometaphase and disassociates upon spindle fiber attachment, indicating that MAD2 regulates kinetochore binding to the spindle fibers. Human MAD2 has also been shown to associate with insulin receptor (IR), but not IGFIR, implicating MAD2 as a mediator for IR-specific signaling. MAD2B, a MAD2 homolog, is required for the execution of the mitotic checkpoint monitoring the kinetochore-spindle attachment process and if the process is not complete, MAD2B delays the onset of anaphase. Applications Suitable for use in Western Blot. Other applications not tested. Recommended Dilution Western Blot: 1:500-1:1000 Optimal dilutions to be determined by the researcher. Storage and Stability May be stored at 4°C for short-term only. Aliquot to avoid repeated freezing and thawing. Store at -20°C. Aliquots are stable for 12 months after receipt. For maximum recovery of product, centrifuge the original vial after thawing and prior to removing the cap. -

Transcriptome Profiling of Human Oocytes Experiencing Recurrent Total
www.nature.com/scientificreports OPEN Transcriptome profling of human oocytes experiencing recurrent total fertilization failure Received: 11 July 2018 Lun Suo1, Yu xiao Zhou2, Li ling Jia2, Hai bo Wu1, Jin Zheng2, Qi feng Lyu1, Li hua Sun1, Accepted: 16 November 2018 Han Sun3 & Yan ping Kuang1 Published: xx xx xxxx There exist some patients who face recurrent total fertilization failure during assisted reproduction treatment, but the pathological mechanism underlying is elusive. Here, by using sc-RNA-seq method, the transcriptome profles of ten abnormally fertilized zygotes were assessed, including fve zygotes from one patient with recurrent Poly-PN zygotes, and fve zygotes from a patient with pronuclear fusion failure. Four zygotes with three pronuclear (Tri-PN) were collected from four diferent patients as controls. After that, we identifed 951 and 1697 signifcantly diferentially expressed genes (SDEGs) in Poly-PN and PN arrest zygotes, respectively as compared with the control group. KEGG analyses indicated down regulated genes in the Poly-PN group included oocyte meiosis related genes, such as PPP2R1B, YWHAZ, MAD2L1, SPDYC, SKP1 and CDC27, together with genes associated with RNA processing, such as SF3B1, LOC645691, MAGOHB, PHF5A, PRPF18, DDX5, THOC1 and BAT1. In contrast, down regulated genes in the PN arrest group, included cell cycle genes, such as E2F4, DBF4, YWHAB, SKP2, CDC23, SMC3, CDC25A, CCND3, BUB1B, MDM2, CCNA2 and CDC7, together with homologous recombination related genes, such as NBN, XRCC3, SHFM1, RAD54B and RAD51. Thus, our work provides a better understanding of transcriptome profles underlying RTFF, although it based on a limited number of patients. Despite nearly forty years of scientifc and clinical advance in the feld of assisted reproduction, there still exist some rarely patients, even though rarely occur, who have to face recurrent total fertilization failure (RTFF) with- out any visual precautionary indicator1–3, even some of them could be rescued by assisted oocyte activation4.