Downloaded from Pubchem in SDF Format
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Design. Concepts and Applications Gisbert Schneider & Karl-Heinz Baringhaus DOI: 10.3395/Reciis.V3i2.259En
[www.reciis.cict.fiocruz.br] ISSN 1981-6286 Book Reviews Molecular design. Concepts and applications Gisbert Schneider & Karl-Heinz Baringhaus DOI: 10.3395/reciis.v3i2.259en Carlos Alberto Manssour Fraga Faculty of Pharmacy, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil [email protected] This book focuses on how different concepts and molecular modeling strategies could be exploited to design the molecular architecture of ligands for specific targets, as new drug candidates. The authors have large experience in drug discovery, occupying head positions in European pharmaceutical industries, which was crucial to their particular form to address the main design-related medicinal chemistry topics along the five chapters of the book. The foreword written by Hugo Kubinyi describes the importance of integrated and multidisciplinary work for the successful of rational drug design process. So, Schneider and Baringhaus used an easy and clear lan- guage to show the main advantages and disadvantages of several different approaches and technologies applied in molecular design of new chemical entities, highlighting the pitfalls and risks of the complex task of discovery a new drug. The first chapter presents the principal structural elements related to the molecules and what features should be persecuted to avoid or minimize the probability of unsuccessful during the clinical trials due to inadequate ADMET properties. In addition to the description of druglikeness properties, an important discussion about the importance of getting informations about the shape and the bioactive conformation of the Wiley-VCH Verlag GmbH, bioligands is included. The second one introduces the Alemanha; 2008 phenomena related to receptor-ligand interactions point- ing the more frequent types of bonds involved in the binding and how the enthalpic and entropic contribu- ISBN: 978-3527314324 tions could benefit the formation of stable complexes. -

Relief of Hypersensitivity After Nerve Injury from Systemic Donepezil
Saranya devi Relief of Hypersensitivity after Nerve Injury from ALN Systemic Donepezil Involves Spinal Cholinergic and γ-Aminobutyric Acid Mechanisms Spinal ACh and GABA Mechanisms for Donepezil Analgesia Masafumi Kimura, M.D.,* Ken-ichiro Hayashida, D.V.M., Ph.D.,† James C. Eisenach, M.D.,‡ KIMURA ET AL. Shigeru Saito M.D.,§ Hideaki Obata, M.D.‖ XX ABSTRACT What We Already Know about This Topic • Donepezil increases central nervous system acetylcholine lev- Downloaded from http://pubs.asahq.org/anesthesiology/article-pdf/118/1/173/259568/0000542-201301000-00030.pdf by guest on 28 September 2021 10.1097/ALN.0b013e318277a81c Background: Evoking spinal release of acetylcholine (ACh) els and is used for treatment of dementia produces antinociception in normal animals and reduces • Increases in spinal acetylcholine levels have been implicated in the relief of neuropathic pain hypersensitivity after nerve injury, and some studies suggest January that ACh-mediated analgesia relies on γ-aminobutyric acid (GABA)-ergic signaling in the spinal cord. In this study, the authors tested the spinal mechanisms underlying the anti- What This Article Tells Us That Is New 118 hypersensitivity effects of donepezil, a central nervous sys- • Donepezil increased spinal acetylcholine levels and γ- tem–penetrating cholinesterase inhibitor, in a rat model of aminobutyric acid levels to reduce nociceptive responses after nerve injury and represents a potential therapeutic pathway to neuropathic pain. reduce pain after nerve injury Methods: Male Sprague-Dawley rats were anesthetized, and L5 spinal nerve ligation was performed unilaterally. With- A antagonist; and CGP 35348 (30 μg), a γ-aminobutyric drawal threshold to a paw pressure test was measured before acid receptor type B antagonist. -

Fast Three Dimensional Pharmacophore Virtual Screening of New Potent Non-Steroid Aromatase Inhibitors
View metadata, citation and similar papers at core.ac.uk brought to you by CORE J. Med. Chem. 2009, 52, 143–150 143 provided by Estudo Geral Fast Three Dimensional Pharmacophore Virtual Screening of New Potent Non-Steroid Aromatase Inhibitors Marco A. C. Neves,† Teresa C. P. Dinis,‡ Giorgio Colombo,*,§ and M. Luisa Sa´ e Melo*,† Centro de Estudos Farmaceˆuticos, Laborato´rio de Quı´mica Farmaceˆutica, Faculdade de Farma´cia, UniVersidade de Coimbra, 3000-295, Coimbra, Portugal, Centro de Neurocieˆncias, Laborato´rio de Bioquı´mica, Faculdade de Farma´cia, UniVersidade de Coimbra, 3000-295, Coimbra, Portugal, and Istituto di Chimica del Riconoscimento Molecolare, CNR, 20131, Milano, Italy ReceiVed July 28, 2008 Suppression of estrogen biosynthesis by aromatase inhibition is an effective approach for the treatment of hormone sensitive breast cancer. Third generation non-steroid aromatase inhibitors have shown important benefits in recent clinical trials with postmenopausal women. In this study we have developed a new ligand- based strategy combining important pharmacophoric and structural features according to the postulated aromatase binding mode, useful for the virtual screening of new potent non-steroid inhibitors. A small subset of promising drug candidates was identified from the large NCI database, and their antiaromatase activity was assessed on an in vitro biochemical assay with aromatase extracted from human term placenta. New potent aromatase inhibitors were discovered to be active in the low nanomolar range, and a common binding mode was proposed. These results confirm the potential of our methodology for a fast in silico high-throughput screening of potent non-steroid aromatase inhibitors. Introduction built and proved to be valuable in understanding the binding 14-16 Aromatase, a member of the cytochrome P450 superfamily determinants of several classes of inhibitors. -

Tor Amendment Tagoor Laboratories Private Limited
ToR Amendment Tagoor Laboratories Private Limited ANNEXURE – I Details of Products as per Granted ToR and Amendment Required Production Capacity S. Ton /Month Therapeutic Product Name No As per Amendment category granted ToR Required 1 Abacavir sulfate 2.0 2.0 Anti HIV 2 Amitriptyline 5.0 10.0 Antidepressant 3 Atrovastatin Calcium 5.0 5.0 Hypercholesterolemia 4 Bupropion 5.0 5.0 Anti depressant 5 carisoprodol 2.0 2.0 Muscle Relaxant 6 Clopidogrelbisulfate 5.0 5.0 Antithrombotic 7 Cyclobenzaprine HCl 5.0 5.0 Muscle relaxant 8 Cyproheptadine HCl 5.0 10.0 Anti allergic 9 Desloratadine 5.0 5.0 Antihistamine 10 Domperidone 20.0 30.0 Anti emetic 11 Domperidone maleate 2.0 2.0 Anti emetic 12 Donepezil HCl 1.0 1.0 Alzheimer’s disease 13 Ebastine 5.0 5.0 Anti allergic 14 Esomeprazole Sodium 3.0 3.0 Anti ulcerative Esomeprazole Magnesium 3.0 3.0 Anti ulcerative 15 trihydrate 16 Fexofenadine Hydrochloride 6.0 15.0 Anti histamine 17 Haloperidol 2.0 2.0 Antipsychotic 18 Itopride Hydrochloride 2.0 2.0 Antispasmodics 19 Itraconazole 10.0 10.0 Antifungal 20 Ketrolac Tromethane 2.0 2.0 Anti Inflammatory 21 Lansoprazole 10.0 10.0 Ant ulcerative 22 Loperamide Hydrochloride 10.0 10.0 Anti diarrhea agent 23 Losartan Potassium 6.0 15.0 Anti Hypertensive 24 Nebivolol HCl 2.0 2.0 Anti Hypertensive 25 Nortriptyline HCl 2.0 2.0 Anti depressant 26 Omeprazole 40.0 40.0 Anti ulcerative 27 Omeprazole Sodium 2.0 2.0 Ant ulcerative Omeprazole Magnesium 2.0 2.0 Ant ulcerative 28 Dihydrate 29 Oxatomide 1.0 1.0 Antihistamine Pantoprazole Sodium Sesqui 10.0 20.0 Ant ulcerative 30 Hydrate 31 Pimozide 2.0 2.0 Antipsychotic 32 Pregabalin 2.0 2.0 Epileptic 33 Quetiapine Hemifumarate 2.0 2.0 Antipsychotic Prepared By Rightsource Industrial Solutions Pvt. -

Downloading Only Compounds with the Properties “Drug-Like”, “Purchasable”
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.02.433618; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Pharmacophore Model for SARS-CoV-2 3CLpro Small Molecule Inhibitors and in Vitro Experimental Validation of Computationally Screened Inhibitors Enrico Glaab*†1, Ganesh Babu Manoharan2, Daniel Abankwa*2 1Luxembourg Centre for Systems Biomedicine (LCSB), University of Luxembourg, 7 avenue des Hauts Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg 2Department of Life Sciences and Medicine, University of Luxembourg, 7 avenue des Hauts Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg *EG and DA are joint senior autHors †Contact: [email protected] Abstract Among the biomedical efforts in response to the current coronavirus (COVID-19) pandemic, pharmacological strategies to reduce viral load in patients with severe forms of the disease are being studied intensively. One of the main drug target proteins proposed so far is the SARS- CoV-2 viral protease 3CLpro (also called Mpro), an essential component for viral replication. Ongoing ligand- and receptor-based computational screening efforts would be facilitated by an improved understanding of the electrostatic, hydrophobic and steric features that characterize small molecule inhibitors binding stably to 3CLpro, as well as by an extended collection of known binders. Here, we present combined virtual screening, molecular dynamics simulation, machine learning and in vitro experimental validation analyses which have led to the identification of small molecule inhibitors of 3CLpro with micromolar activity, and to a pharmacophore model that describes functional chemical groups associated with the molecular recognition of ligands by the 3CLpro binding pocket. -
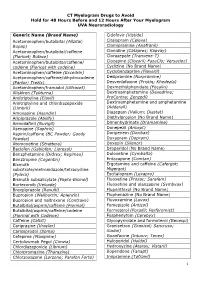
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology Generic Name (Brand Name) Cidofovir (Vistide) Acetaminophen/butalbital (Allzital; Citalopram (Celexa) Bupap) Clomipramine (Anafranil) Acetaminophen/butalbital/caffeine Clonidine (Catapres; Kapvay) (Fioricet; Butace) Clorazepate (Tranxene-T) Acetaminophen/butalbital/caffeine/ Clozapine (Clozaril; FazaClo; Versacloz) codeine (Fioricet with codeine) Cyclizine (No Brand Name) Acetaminophen/caffeine (Excedrin) Cyclobenzaprine (Flexeril) Acetaminophen/caffeine/dihydrocodeine Desipramine (Norpramine) (Panlor; Trezix) Desvenlafaxine (Pristiq; Khedezla) Acetaminophen/tramadol (Ultracet) Dexmethylphenidate (Focalin) Aliskiren (Tekturna) Dextroamphetamine (Dexedrine; Amitriptyline (Elavil) ProCentra; Zenzedi) Amitriptyline and chlordiazepoxide Dextroamphetamine and amphetamine (Limbril) (Adderall) Amoxapine (Asendin) Diazepam (Valium; Diastat) Aripiprazole (Abilify) Diethylpropion (No Brand Name) Armodafinil (Nuvigil) Dimenhydrinate (Dramamine) Asenapine (Saphris) Donepezil (Aricept) Aspirin/caffeine (BC Powder; Goody Doripenem (Doribax) Powder) Doxapram (Dopram) Atomoxetine (Strattera) Doxepin (Silenor) Baclofen (Gablofen; Lioresal) Droperidol (No Brand Name) Benzphetamine (Didrex; Regimex) Duloxetine (Cymbalta) Benztropine (Cogentin) Entacapone (Comtan) Bismuth Ergotamine and caffeine (Cafergot; subcitrate/metronidazole/tetracycline Migergot) (Pylera) Escitalopram (Lexapro) Bismuth subsalicylate (Pepto-Bismol) Fluoxetine (Prozac; Sarafem) -

The Selection & Application of Free Prediction Models for Drug Discovery
The selection & application of free prediction models for drug discovery • Scope • Sources of in silico models • Assessing model quality in series optimisation • Model quality in general • Models & HTS triage • Lipophilicity • Tautomers Disclaimer: There are lots of opinions on this topic, these are ours…. With many thanks to Caroline Low & Alexander Alex 1 Scope • Models of pharmacokinetics & toxicity • Activity models, docking and scoring are not explicitly considered though many of the principles will be applicable • A selection of models & tools, the list is not exhaustive specific models general ‘models’ brain penetration lipophilicity druglikeness solubility pKa toxicity metabolism absorption/efflux 2 Models, models, models octanol water metabolism logP kinetic solubility cytotoxicity • We use a multitude of models in drug discovery • Many are experimentally derived ‘surrogate endpoints’ that we hope will correlate with something useful in human • in silico models are often predictions of the surrogate endpoints 3 Example software & in silico models (not an exhaustive list) DataWarrior1 swissADME2 ChemBench3 StarDrop4 chembench.mml.unc.edu/ openmolecules.org swissadme.ch prediction.action optibrium.com • logP • logP • 125 models • logP • solubility • 6 methods including: • logD7.4 • solubility • BBB • solubility • 3 methods • tox (various) • aq • absorption • PPB • PBS pH7.4 • GI • skin • HIA • BBB permeability • BBB • skin • Transporters • P450 affinities • Pgp • Pgp • Cyps (5) • hERG • PPB 1. Free 2. Free but require users to -

Retrospective Observation of Liver Function Parameters for 101 Patients Using Herbal Drugs for One Month
31 2 (2010 3 ) J Korean Oriental Med 2010;31(2):149-157 Original article 김동민1, 김회권2, 조성연3, 김용석1, 남상수1 1경희대학교 한의과대학 침구학교실, 2가평군 보건소, 3경희대학교 강남경희한방병원 침구과 Retrospective Observation of Liver Function Parameters for 101 Patients Using Herbal Drugs for One Month Dong-Min Kim 1, Hyee-Kwon Kim 2, Seong-Yeun Cho 3, Yong-Suk Kim 1, Sang-Soo Nam 1 1Dept. of Acupuncture & Moxibustion, College of Oriental Medicine, Kyung Hee University 2Gapyeong Public Health Center 3Dept. of Acupuncture & Moxibustion, Kangnam KyungHee Korean Hospital, Kyung Hee University Objective: The purpose of this study was to investigate the level of safety on liver functions when Korean herbal medicine was taken internally. Method: 101 inpatients who took Korean herbal medicine were enrolled and liver function test (aspartic aminotransferase, alanine aminotransferase, alkaline phosphatase, total bilirubin, gamma glutamyltranspeptidase, albumin, lactate dehydrogenase) was performed on admission and 1 month later. Results: In 101 patients, alkaline phosphatase and gamma glutamyltranspeptidase decreased significantly compared with the value taken on admission (p<0.05) but aspartic aminotransferase, alanine aminotransferase, total bilirubin, albumin, and lactate dehydrogenase were not significantly changed (p>0.05). In the patients who took Scutellaria baicalensis (n=34), alkaline phosphatase decreased and albumin increased significantly (p<0.05). Among the patients who took Atractylodes macrocephala (n=29), alkaline phosphatase decreased significantly (p<0.05). In the patients who took Glycyrrhiza uralensis or Paeonia lactiflora , liver function parameters were not significantly changed (p>0.05). On admission 11 patients had abnormal liver function and 2 patients had liver injury while 7 patients had abnormal liver function and 2 patients showed liver injury 1 month later. -

Overselling Donepezil (ARICEPD and Exploiting Patients V1ith I\Lzheimers Disease: Why Isn't the FDA Stopping These Ads?
PUBLIC CITIZEN HEALTH RESEARCH GROUP SIDNEY M. WOLFE, M.D., EDITOR March 2004 + VOL. 20, N0 . 3 Overselling Donepezil (ARICEPD and Exploiting Patients v1ith i\lzheimers Disease: Why Isn't the FDA Stopping These Ads? n the 1999 Edition of Worst Pills, patients. The ads proclaim that If this conclusion were based on a Best Pills, we urged patients not patients with Alzheimer's disease randomized, placebo-controlled trial I to use donepezil (ARICEPT), who "persistently" use donepezil are (RCT) (the scientific gold standard for made by Pfizer/ Esai, because of a able to stay in the community and medical research) wherein 50 percent combination of adverse effects such avoid going into a nursing home for of the patients got donepezil and the as nausea, diarrhea and vomiting almost two years more than patients other 50 percent of exactly the same along with serious questions about who have only limited treatment or kinds of patients were randomly clinically significant effectiveness. no treatment with the dmg (see assigned to be given a placebo, it We quoted the prestigious Medical advertisement on page 2 for an would be a major public health Letter, an independent evaluation for example). The implication is star breakthrough and a cause for cele physicians of newly approved dmgs, tling. The families and other friends bration. Unfortunately, it is not based which stated "There is no evidence who are caring for patients with on such a study and is not any cause that use of... donepezil... leads to Alzheimer's disease will have a for celebration. substantial functional improvement substantial amount of extra time to The study cited as the basis for this or prevents the progression of the have their loved ones in the commu seemingly remarkable improvement disease." Since then, Pfizer/ Esai has nity before they have to go into a is flawed. -

Drugs Affectin the Autonomic Nervous System
Fundamentals of Medical Pharmacology Paterson Public Schools Written by Néstor Collazo, Ph.D. Jonathan Hodges, M.D. Tatiana Mikhaelovsky, M.D. for Health and Related Professions (H.A.R.P.) Academy March 2007 Course Description This fourth year course is designed to give students in the Health and Related Professions (H.A.R.P.) Academy a general and coherent explanation of the science of pharmacology in terms of its basic concepts and principles. Students will learn the properties and interactions between chemical agents (drugs) and living organisms for the rational and safe use of drugs in the control, prevention, and therapy of human disease. The emphasis will be on the fundamental concepts as they apply to the actions of most prototype drugs. In order to exemplify important underlying principles, many of the agents in current use will be singled out for fuller discussion. The course will include the following topics: ¾ The History of Pharmacology ¾ Terminology Used in Pharmacology ¾ Drug Action on Living Organisms ¾ Principles of Pharmacokinetics ¾ Dose-Response Relationships ¾ Time-Response Relationships ¾ Human Variability: Factors that will modify effects of drugs on individuals ¾ Effects of Drugs Attributable to Varying Modes of Administration ¾ Drug Toxicity ¾ Pharmacologic Aspects of Drug Abuse and Drug Dependence Pre-requisites Students must have completed successfully the following courses: Biology, Chemistry, Anatomy and Physiology, Algebra I and II Credits: 5 credits Basic Principles of Drug Action Introduction to Pharmacology a. Basic Mechanisms of Drug Actions b. Dose-response relationships c. Drug absorption d. Biotransformation of Drugs e. Pharmacokinetics f. Factors Affecting Drug Distribution g. Drug Allergy and Pharmacogenetics h. -

Potential Herb–Drug Interactions in the Management of Age-Related Cognitive Dysfunction
pharmaceutics Review Potential Herb–Drug Interactions in the Management of Age-Related Cognitive Dysfunction Maria D. Auxtero 1, Susana Chalante 1,Mário R. Abade 1 , Rui Jorge 1,2,3 and Ana I. Fernandes 1,* 1 CiiEM, Interdisciplinary Research Centre Egas Moniz, Instituto Universitário Egas Moniz, Quinta da Granja, Monte de Caparica, 2829-511 Caparica, Portugal; [email protected] (M.D.A.); [email protected] (S.C.); [email protected] (M.R.A.); [email protected] (R.J.) 2 Polytechnic Institute of Santarém, School of Agriculture, Quinta do Galinheiro, 2001-904 Santarém, Portugal 3 CIEQV, Life Quality Research Centre, IPSantarém/IPLeiria, Avenida Dr. Mário Soares, 110, 2040-413 Rio Maior, Portugal * Correspondence: [email protected]; Tel.: +35-12-1294-6823 Abstract: Late-life mild cognitive impairment and dementia represent a significant burden on health- care systems and a unique challenge to medicine due to the currently limited treatment options. Plant phytochemicals have been considered in alternative, or complementary, prevention and treat- ment strategies. Herbals are consumed as such, or as food supplements, whose consumption has recently increased. However, these products are not exempt from adverse effects and pharmaco- logical interactions, presenting a special risk in aged, polymedicated individuals. Understanding pharmacokinetic and pharmacodynamic interactions is warranted to avoid undesirable adverse drug reactions, which may result in unwanted side-effects or therapeutic failure. The present study reviews the potential interactions between selected bioactive compounds (170) used by seniors for cognitive enhancement and representative drugs of 10 pharmacotherapeutic classes commonly prescribed to the middle-aged adults, often multimorbid and polymedicated, to anticipate and prevent risks arising from their co-administration. -

Donepezil, a Drug for Alzheimer’S Disease, Promotes Oligodendrocyte
www.nature.com/aps ARTICLE Donepezil, a drug for Alzheimer’s disease, promotes oligodendrocyte generation and remyelination Xue Cui1, Yu-e Guo2,3, Jia-hui Fang1, Chang-jie Shi1, Na Suo2,3, Ru Zhang1 and Xin Xie1,2,4 Myelin sheaths play important roles in neuronal functions. In the central nervous system (CNS), the myelin is formed by oligodendrocytes (OLs), which are differentiated from oligodendrocyte precursor cells (OPCs). In CNS demyelinating disorders such as multiple sclerosis (MS), the myelin sheaths are damaged and the remyelination process is hindered. Small molecule drugs that promote OPC to OL differentiation and remyelination may provide a new way to treat these demyelinating diseases. Here we report that donepezil, an acetylcholinesterase inhibitor (AChEI) developed for the treatment of Alzheimer’s disease (AD), significantly promotes OPC to OL differentiation. Interestingly, other AChEIs, including huperzine A, rivastigmine, and tacrine, have no such effect, indicating that donepezil’s effect in promoting OPC differentiation is not dependent on the inhibition of AChE. Donepezil also facilitates the formation of myelin sheaths in OPC–DRG neuron co-culture. More interestingly, donepezil also promotes the repair of the myelin sheaths in vivo and provides significant therapeutic effect in a cuprizone-mediated demyelination animal model. Donepezil is a drug that has been used to treat AD safely for many years; our findings suggest that it might be repurposed to treat CNS demyelinating diseases such as MS by promoting OPC to OL differentiation and remyelination. Keywords: donepezil; aricept; oligodendrocyte; oligodendrocyte progenitor cell; differentiation; myelin sheaths; remyelination; CNS demyelinating disorders; multiple sclerosis Acta Pharmacologica Sinica (2019) 40:1386–1393; https://doi.org/10.1038/s41401-018-0206-4 INTRODUCTION mitochondria function, has been found to accelerate OL matura- Oligodendrocytes (OLs), an important component of neuroglia, tion in culture and increase remyelination in rodents [11, 12].