Dendritic Cell Development and Function
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

List of Genes Used in Cell Type Enrichment Analysis
List of genes used in cell type enrichment analysis Metagene Cell type Immunity ADAM28 Activated B cell Adaptive CD180 Activated B cell Adaptive CD79B Activated B cell Adaptive BLK Activated B cell Adaptive CD19 Activated B cell Adaptive MS4A1 Activated B cell Adaptive TNFRSF17 Activated B cell Adaptive IGHM Activated B cell Adaptive GNG7 Activated B cell Adaptive MICAL3 Activated B cell Adaptive SPIB Activated B cell Adaptive HLA-DOB Activated B cell Adaptive IGKC Activated B cell Adaptive PNOC Activated B cell Adaptive FCRL2 Activated B cell Adaptive BACH2 Activated B cell Adaptive CR2 Activated B cell Adaptive TCL1A Activated B cell Adaptive AKNA Activated B cell Adaptive ARHGAP25 Activated B cell Adaptive CCL21 Activated B cell Adaptive CD27 Activated B cell Adaptive CD38 Activated B cell Adaptive CLEC17A Activated B cell Adaptive CLEC9A Activated B cell Adaptive CLECL1 Activated B cell Adaptive AIM2 Activated CD4 T cell Adaptive BIRC3 Activated CD4 T cell Adaptive BRIP1 Activated CD4 T cell Adaptive CCL20 Activated CD4 T cell Adaptive CCL4 Activated CD4 T cell Adaptive CCL5 Activated CD4 T cell Adaptive CCNB1 Activated CD4 T cell Adaptive CCR7 Activated CD4 T cell Adaptive DUSP2 Activated CD4 T cell Adaptive ESCO2 Activated CD4 T cell Adaptive ETS1 Activated CD4 T cell Adaptive EXO1 Activated CD4 T cell Adaptive EXOC6 Activated CD4 T cell Adaptive IARS Activated CD4 T cell Adaptive ITK Activated CD4 T cell Adaptive KIF11 Activated CD4 T cell Adaptive KNTC1 Activated CD4 T cell Adaptive NUF2 Activated CD4 T cell Adaptive PRC1 Activated -

Single-Cell RNA Sequencing Demonstrates the Molecular and Cellular Reprogramming of Metastatic Lung Adenocarcinoma
ARTICLE https://doi.org/10.1038/s41467-020-16164-1 OPEN Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma Nayoung Kim 1,2,3,13, Hong Kwan Kim4,13, Kyungjong Lee 5,13, Yourae Hong 1,6, Jong Ho Cho4, Jung Won Choi7, Jung-Il Lee7, Yeon-Lim Suh8,BoMiKu9, Hye Hyeon Eum 1,2,3, Soyean Choi 1, Yoon-La Choi6,10,11, Je-Gun Joung1, Woong-Yang Park 1,2,6, Hyun Ae Jung12, Jong-Mu Sun12, Se-Hoon Lee12, ✉ ✉ Jin Seok Ahn12, Keunchil Park12, Myung-Ju Ahn 12 & Hae-Ock Lee 1,2,3,6 1234567890():,; Advanced metastatic cancer poses utmost clinical challenges and may present molecular and cellular features distinct from an early-stage cancer. Herein, we present single-cell tran- scriptome profiling of metastatic lung adenocarcinoma, the most prevalent histological lung cancer type diagnosed at stage IV in over 40% of all cases. From 208,506 cells populating the normal tissues or early to metastatic stage cancer in 44 patients, we identify a cancer cell subtype deviating from the normal differentiation trajectory and dominating the metastatic stage. In all stages, the stromal and immune cell dynamics reveal ontological and functional changes that create a pro-tumoral and immunosuppressive microenvironment. Normal resident myeloid cell populations are gradually replaced with monocyte-derived macrophages and dendritic cells, along with T-cell exhaustion. This extensive single-cell analysis enhances our understanding of molecular and cellular dynamics in metastatic lung cancer and reveals potential diagnostic and therapeutic targets in cancer-microenvironment interactions. 1 Samsung Genome Institute, Samsung Medical Center, Seoul 06351, Korea. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -

Human CLEC4C / BDCA2 ELISA Kit (ARG82536)
Product datasheet [email protected] ARG82536 Package: 96 wells Human CLEC4C / BDCA2 ELISA Kit Store at: 4°C Component Cat. No. Component Name Package Temp ARG82536-001 Antibody-coated 8 X 12 strips 4°C. Unused strips microplate should be sealed tightly in the air-tight pouch. ARG82536-002 Standard 2 X 10 ng/vial 4°C ARG82536-003 Standard/Sample 30 ml (Ready to use) 4°C diluent ARG82536-004 Antibody conjugate 1 vial (100 µl) 4°C concentrate (100X) ARG82536-005 Antibody diluent 12 ml (Ready to use) 4°C buffer ARG82536-006 HRP-Streptavidin 1 vial (100 µl) 4°C concentrate (100X) ARG82536-007 HRP-Streptavidin 12 ml (Ready to use) 4°C diluent buffer ARG82536-008 25X Wash buffer 20 ml 4°C ARG82536-009 TMB substrate 10 ml (Ready to use) 4°C (Protect from light) ARG82536-010 STOP solution 10 ml (Ready to use) 4°C ARG82536-011 Plate sealer 4 strips Room temperature Summary Product Description ARG82536 Human CLEC4C / BDCA2 ELISA Kit is an Enzyme Immunoassay kit for the quantification of Human CLEC4C / BDCA2 in serum, plasma (EDTA, heparin, citrate) and cell culture supernatants. Tested Reactivity Hu Tested Application ELISA Target Name CLEC4C / BDCA2 Conjugation HRP Conjugation Note Substrate: TMB and read at 450 nm. Sensitivity 10 pg/ml Sample Type Serum, plasma (EDTA, heparin, citrate) and cell culture supernatants. Standard Range 15.6 - 1000 pg/ml Sample Volume 100 µl Precision Intra-Assay CV: 4.4% Inter-Assay CV: 6.7% www.arigobio.com 1/3 Alternate Names CD antigen CD303; BDCA-2; Blood dendritic cell antigen 2; Dendritic lectin; PRO34150; DLEC; C-type lectin domain family 4 member C; CLECSF7; CD303; CLECSF11; HECL; BDCA2; C-type lectin superfamily member 7 Application Instructions Assay Time ~ 5 hours Properties Form 96 well Storage instruction Store the kit at 2-8°C. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Adipose Recruitment and Activation of Plasmacytoid Dendritic Cells Fuel Metaflammation’
Diabetes Page 2 of 61 Adipose recruitment and activation of plasmacytoid dendritic cells fuel metaflammation Amrit Raj Ghosh1, Roopkatha Bhattacharya1, Shamik Bhattacharya1, Titli Nargis2, Oindrila Rahaman1, Pritam Duttagupta1, Deblina Raychaudhuri1, Chinky Shiu Chen Liu1, Shounak Roy1, Parasar Ghosh3, Shashi Khanna4, Tamonas Chaudhuri4, Om Tantia4, Stefan Haak5, Santu Bandyopadhyay1, Satinath Mukhopadhyay6, Partha Chakrabarti2 and Dipyaman Ganguly1*. Divisions of 1Cancer Biology & Inflammatory Disorders and 2Cell Biology & Physiology, CSIR- Indian Institute of Chemical Biology, Kolkata, India; 4ILS Hospitals, Kolkata, India; 5Zentrum Allergie & Umwelt (ZAUM), Technical University of Munich and Helmholtz Centre Munich, Munich, Germany; Departments of 3Rheumatology and 6Endocrinology, Institute of Postgraduate Medical Education & Research, Kolkata, India. *Corresponding author: Dipyaman Ganguly, Division of Cancer Biology & Inflammatory Disorders, CSIR-Indian Institute of Chemical Biology, 4 Raja S C Mullick Road, Jadavpur, Kolkata, West Bengal, India, 700032. Phone: 91 33 24730492 Fax: 91 33 2473 5197 Email: [email protected] Running title: PDCs and type I interferons in metaflammation Word count (Main text): 5521 Figures: 7, Table: 1 1 Diabetes Publish Ahead of Print, published online August 25, 2016 Page 3 of 61 Diabetes ABSTRACT In obese individuals the visceral adipose tissue (VAT) becomes seat of chronic low grade inflammation (metaflammation). But the mechanistic link between increased adiposity and metaflammation remains largely -

KRAS Mutations Are Negatively Correlated with Immunity in Colon Cancer
www.aging-us.com AGING 2021, Vol. 13, No. 1 Research Paper KRAS mutations are negatively correlated with immunity in colon cancer Xiaorui Fu1,2,*, Xinyi Wang1,2,*, Jinzhong Duanmu1, Taiyuan Li1, Qunguang Jiang1 1Department of Gastrointestinal Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People's Republic of China 2Queen Mary College, Medical Department, Nanchang University, Nanchang, Jiangxi, People's Republic of China *Equal contribution Correspondence to: Qunguang Jiang; email: [email protected] Keywords: KRAS mutations, immunity, colon cancer, tumor-infiltrating immune cells, inflammation Received: March 27, 2020 Accepted: October 8, 2020 Published: November 26, 2020 Copyright: © 2020 Fu et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The heterogeneity of colon cancer tumors suggests that therapeutics targeting specific molecules may be effective in only a few patients. It is therefore necessary to explore gene mutations in colon cancer. In this study, we obtained colon cancer samples from The Cancer Genome Atlas, and the International Cancer Genome Consortium. We evaluated the landscape of somatic mutations in colon cancer and found that KRAS mutations, particularly rs121913529, were frequent and had prognostic value. Using ESTIMATE analysis, we observed that the KRAS-mutated group had higher tumor purity, lower immune score, and lower stromal score than the wild- type group. Through single-sample Gene Set Enrichment Analysis and Gene Set Enrichment Analysis, we found that KRAS mutations negatively correlated with enrichment levels of tumor infiltrating lymphocytes, inflammation, and cytolytic activities. -

Immune Suppressive Landscape in the Human Esophageal Squamous Cell
ARTICLE https://doi.org/10.1038/s41467-020-20019-0 OPEN Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment ✉ Yingxia Zheng 1,2,10 , Zheyi Chen1,10, Yichao Han 3,10, Li Han1,10, Xin Zou4, Bingqian Zhou1, Rui Hu5, Jie Hao4, Shihao Bai4, Haibo Xiao5, Wei Vivian Li6, Alex Bueker7, Yanhui Ma1, Guohua Xie1, Junyao Yang1, ✉ ✉ ✉ Shiyu Chen1, Hecheng Li 3 , Jian Cao 7,8 & Lisong Shen 1,9 1234567890():,; Cancer immunotherapy has revolutionized cancer treatment, and it relies heavily on the comprehensive understanding of the immune landscape of the tumor microenvironment (TME). Here, we obtain a detailed immune cell atlas of esophageal squamous cell carcinoma (ESCC) at single-cell resolution. Exhausted T and NK cells, regulatory T cells (Tregs), alternatively activated macrophages and tolerogenic dendritic cells are dominant in the TME. Transcriptional profiling coupled with T cell receptor (TCR) sequencing reveal lineage con- nections in T cell populations. CD8 T cells show continuous progression from pre-exhausted to exhausted T cells. While exhausted CD4, CD8 T and NK cells are major proliferative cell components in the TME, the crosstalk between macrophages and Tregs contributes to potential immunosuppression in the TME. Our results indicate several immunosuppressive mechanisms that may be simultaneously responsible for the failure of immuno-surveillance. Specific targeting of these immunosuppressive pathways may reactivate anti-tumor immune responses in ESCC. 1 Department of Laboratory Medicine, Xin Hua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 2 Institute of Biliary Tract Diseases Research, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 3 Department of Thoracic Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. -

CLEC4C P.K210del Variant Causes Impaired Cell Surface Transport in Plasmacytoid Dendritic Cells of Amyotrophic Lateral Sclerosis
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 18 CLEC4C p.K210del variant causes impaired cell surface transport in plasmacytoid dendritic cells of amyotrophic lateral sclerosis Su Min Lim1,5,*, Young-Eun Kim2,*, Won Jun Choi3, Ki-Wook Oh4,5, Min-Young Noh5, Min-Soo Kwon6, Minyeop Nahm5, Namshin Kim7, Chang-Seok Ki8,*, Seung Hyun Kim1,4,5,* 1 Department of Translational Medicine, Graduate School of Biomedical Science and Engineering, Hanyang University, Seoul, Republic of Korea 2 Green Cross Genome, Yongin, Republic of Korea 3 Department of Neurology, Sheikh Khalifa Specialty Hospital, Ras Al Khaimah, United Arab Emirates 4 Department of Neurology, College of Medicine, Hanyang University, Seoul, Republic of Korea 5 Cell Therapy Center, Hanyang University Hospital, Seoul, Republic of Korea 6 Department of Pharmacology, School of Medicine, CHA University, CHA Bio Complex, Seongnam, Republic of Korea 7 Epigenomics Research Center Genome Institute, Korea Research Institute of Bioscience and Biotechnology, Daejeon, Republic of Korea 8 Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea *These authors contributed equally to this work Correspondence to: Seung Hyun Kim, email: [email protected] Chang-Seok Ki, email: [email protected] Keywords: C-type lectin, whole-exome sequencing, dilysine motif, ER retention, amyotrophic lateral sclerosis, Gerotarget Received: October 07, 2015 Accepted: January 29, 2016 Published: March 01, 2016 ABSTRACT The type II C-type lectin CLEC4C is a transmembrane protein selectively expressed on plasmacytoid dendritic cells (PDCs). Although its mechanism of action remains unclear, triggering of the extracellular C-terminal C-type carbohydrate recognition region of CLEC4C regulates the secretion of proinflammatory cytokines and type I IFNs in PDCs. -

Human CD Marker Chart Reviewed by HLDA1 Bdbiosciences.Com/Cdmarkers
BD Biosciences Human CD Marker Chart Reviewed by HLDA1 bdbiosciences.com/cdmarkers 23-12399-01 CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD1a R4, T6, Leu6, HTA1 b-2-Microglobulin, CD74 + + + – + – – – CD93 C1QR1,C1qRP, MXRA4, C1qR(P), Dj737e23.1, GR11 – – – – – + + – – + – CD220 Insulin receptor (INSR), IR Insulin, IGF-2 + + + + + + + + + Insulin-like growth factor 1 receptor (IGF1R), IGF-1R, type I IGF receptor (IGF-IR), CD1b R1, T6m Leu6 b-2-Microglobulin + + + – + – – – CD94 KLRD1, Kp43 HLA class I, NKG2-A, p39 + – + – – – – – – CD221 Insulin-like growth factor 1 (IGF-I), IGF-II, Insulin JTK13 + + + + + + + + + CD1c M241, R7, T6, Leu6, BDCA1 b-2-Microglobulin + + + – + – – – CD178, FASLG, APO-1, FAS, TNFRSF6, CD95L, APT1LG1, APT1, FAS1, FASTM, CD95 CD178 (Fas ligand) + + + + + – – IGF-II, TGF-b latency-associated peptide (LAP), Proliferin, Prorenin, Plasminogen, ALPS1A, TNFSF6, FASL Cation-independent mannose-6-phosphate receptor (M6P-R, CIM6PR, CIMPR, CI- CD1d R3G1, R3 b-2-Microglobulin, MHC II CD222 Leukemia -
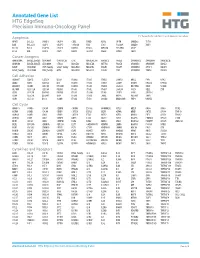
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1