Role of Myeloid-Derived Suppressor Cells in Tumor-Associated Pregnancy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Types of Acute Phase Reactants and Their Importance in Vaccination (Review)
BIOMEDICAL REPORTS 12: 143-152, 2020 Types of acute phase reactants and their importance in vaccination (Review) RAFAAT H. KHALIL1 and NABIL AL-HUMADI2 1Department of Biology, College of Science and Technology, Florida Agricultural and Mechanical University, Tallahassee, FL 32307; 2Office of Vaccines, Food and Drug Administration, Center for Biologics Evaluation and Research, Silver Spring, MD 20993, USA Received May 10, 2019; Accepted November 25, 2019 DOI: 10.3892/br.2020.1276 Abstract. Vaccines are considered to be one of the most human and veterinary medicine. Proteins which are expressed cost-effective life-saving interventions in human history. in the acute phase are potential biomarkers for the diagnosis The body's inflammatory response to vaccines has both of inflammatory disease, for example, acute phase proteins desired effects (immune response), undesired effects [(acute (APPs) are indicators of successful organ transplantation phase reactions (APRs)] and trade‑offs. Trade‑offs are and can be used to predict the ameliorative effect of cancer more potent immune responses which may be potentially therapy (1,2). APPs are primarily synthesized in hepatocytes. difficult to separate from potent acute phase reactions. The acute phase response is a spontaneous reaction triggered Thus, studying acute phase proteins (APPs) during vaccina- by disrupted homeostasis resulting from environmental distur- tion may aid our understanding of APRs and homeostatic bances (3). Acute phase reactions (APRs) usually stabilize changes which can result from inflammatory responses. quickly, after recovering from a disruption to homeostasis Depending on the severity of the response in humans, these within a few days to weeks; however, APPs expression levels reactions can be classified as major, moderate or minor. -

Influence of Infection and Inflammation on Biomarkers of Nutritional Status
A2.4 INFLUENCE OF INFECTION AND INFLAMMATION ON BIOMARKERS OF NUTRITIONAL STATUS A2.4 Influence of infection and inflammation on biomarkers of nutritional status with an emphasis on vitamin A and iron David I. Thurnham1 and George P. McCabe2 1 Northern Ireland Centre for Food and Health, University of Ulster, Coleraine, United Kingdom of Great Britain and Northern Ireland 2 Statistics Department, Purdue University, West Lafayette, Indiana, United States of America Corresponding author: David I. Thurnham; [email protected] Suggested citation: Thurnham DI, McCabe GP. Influence of infection and inflammation on biomarkers of nutritional status with an emphasis on vitamin A and iron. In: World Health Organization. Report: Priorities in the assessment of vitamin A and iron status in populations, Panama City, Panama, 15–17 September 2010. Geneva, World Health Organization, 2012. Abstract n Many plasma nutrients are influenced by infection or tissue damage. These effects may be passive and the result of changes in blood volume and capillary permeability. They may also be the direct effect of metabolic alterations that depress or increase the concentration of a nutrient or metabolite in the plasma. Where the nutrient or metabolite is a nutritional biomarker as in the case of plasma retinol, a depression in retinol concentrations will result in an overestimate of vitamin A deficiency. In contrast, where the biomarker is increased due to infection as in the case of plasma ferritin concentrations, inflammation will result in an underestimate of iron deficiency. Infection and tissue damage can be recognized by their clinical effects on the body but, unfortunately, subclinical infection or inflammation can only be recognized by measur- ing inflammation biomarkers in the blood. -

LEUKOCYTE SURFACE ORIGIN of HUMAN At-ACID GLYCOPROTEIN (OROSOMUCOID)*
LEUKOCYTE SURFACE ORIGIN OF HUMAN at-ACID GLYCOPROTEIN (OROSOMUCOID)* BY CARL G. GAHMBERG AND LEIF C. ANDERSSON (From the Department of Bacteriology and Immunology, and the Transplantation Laboratory, Department of Surgery IV, University of Helsinki, Helsinki 29, Finland) Human al-acid glycoprotein (orosomucoid) (o~I-AG)1 constitutes the main component of the seromucoid fraction of human plasma. It belongs to the acute phase proteins, which increase under conditions such as inflammation, pregnancy, and cancer (1, 2). al-AG has previously been found to be synthesized in liver (3), and after removal of terminal sialic acids, it is cleared from the circulation by binding to a receptor protein on liver cell plasma membranes (4). The structure of al-AG is well known. It is composed of a single polypeptide chain and contains 245% carbohydrate including a large amount of sialic acid. The carbohydrate is located in the first half of the peptide chain linked to asparagine residues (5, 6). The function of al-AG is unclear. However, Schmid et al. (5) and Ikenaka et al. (7) and reported that the amino acid sequence of the protein shows a significant homology with human IgG. This finding and the striking increase in inflammatory and lymphopro- liferative disorders made us consider the possibility that leukocytes could be directly involved in the synthesis and release of a~-AG. We report here the presence of a membrane form of al-AG, with an apparent tool wt of 52,000, on normal human lymphocytes, granulocytes, and monocytes. By the use of internal labeling with [3H]leucine in vitro, we demonstrate that the membrane protein is synthesized by lymphocytes. -

Downloaded from Bioscientifica.Com at 09/25/2021 07:25:24AM Via Free Access 812 M Andreassen and Others EUROPEAN JOURNAL of ENDOCRINOLOGY (2012) 166
European Journal of Endocrinology (2012) 166 811–819 ISSN 0804-4643 CLINICAL STUDY GH activity and markers of inflammation: a crossover study in healthy volunteers treated with GH and a GH receptor antagonist Mikkel Andreassen1, Jan Frystyk2,3, Jens Faber1,4 and Lars Østergaard Kristensen1 1Endocrine Unit, Laboratory of Endocrinology 54o4, Department of Internal Medicine O, Herlev Hospital, University of Copenhagen, Herlev Ringvej 75, DK-2730 Herlev, Denmark, 2Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Aarhus, Denmark and 3Medical Research Laboratories, Faculty of Health Sciences, Institute of Clinical Medicine, Aarhus University, Aarhus, Denmark and 4Faculty of Health Science, Copenhagen University, Copenhagen, Denmark (Correspondence should be addressed to M Andreassen; Email: [email protected]) Abstract Introduction: The GH/IGF1 axis may modulate inflammatory processes. However, the relationship seems complicated as both pro- and anti-inflammatory effects have been demonstrated. Methods/design: Twelve healthy volunteers (mean age 36, range 27–49 years) were treated in random order with increasing doses of GH for 3 weeks (first week 0.01 mg/kg per day, second week 0.02 mg/kg per day, and third week 0.03 mg/kg per day) or a GH receptor antagonist (pegvisomant; first week 10 mg/day and last two weeks 15 mg/day), separated by 8 weeks of washout. Circulating levels of the pro-inflammatory cytokines tumor necrosis factor a (TNFa (TNFA)), interleukin 6 (IL6), and IL1b (IL1B) and the acute phase proteins (APPs) C-reactive protein (CRP), haptoglobin, orosomucoid, YKL40 (CHI3L1), and fibrinogen were measured. Results: During GH treatment, IGF1 (median 131 (Inter-quartile range (IQR) 112–166) vs 390 (322– 524) mg/l, PZ0.002) increased together with TNFa (0.87 (0.74–1.48) vs 1.27 (0.80–1.69) ng/l, PZ0.003), IL6 (1.00 (0.83–1.55) vs 1.35 (0.80–4.28) ng/l, PZ0.045), and fibrinogen (9.2 (8.8–9.6) vs 11.1 (9.4–12.4) mM, PZ0.002). -

The Acute-Phase Protein Orosomucoid Regulates Food Intake and Energy Homeostasis Via Leptin Receptor Signaling Pathway
1630 Diabetes Volume 65, June 2016 Yang Sun,1 Yili Yang,2 Zhen Qin,1 Jinya Cai,3 Xiuming Guo,1 Yun Tang,3 Jingjing Wan,1 Ding-Feng Su,1 and Xia Liu1 The Acute-Phase Protein Orosomucoid Regulates Food Intake and Energy Homeostasis via Leptin Receptor Signaling Pathway Diabetes 2016;65:1630–1641 | DOI: 10.2337/db15-1193 The acute-phase protein orosomucoid (ORM) exhibits a intake and energy expenditure. Energy homeostasis in the variety of activities in vitro and in vivo, notably modulation body is maintained by the integrated actions of multiple of immunity and transportation of drugs. We found in this factors (1,2), including adipose hormones (such as leptin study that mice lacking ORM1 displayed aberrant energy and adiponectin), gastrointestinal hormones (such as in- homeostasis characterized by increased body weight and sulin, ghrelin, and cholecystokinin), and nutrient-related fat mass. Further investigation found that ORM, predom- signals (such as free fatty acids). In addition to acting on fi inantly ORM1, is signi cantly elevated in sera, liver, and peripheral tissues, these actions can also influence central – adipose tissues from the mice with high-fat diet (HFD) circuits in the hypothalamus, brainstem, and limbic system db/db induced obesity and mice that develop obesity to modulate food intake and energy expenditure (1,3). spontaneously due to mutation in the leptin receptor Notably, the adipose tissue–produced leptin is a major (LepR). Intravenous or intraperitoneal administration of regulator of fat, and the level of leptin in circulation is exogenous ORM decreased food intake in C57BL/6, HFD, proportional to body fat (4) and is a reflection of long- and leptin-deficient ob/ob mice, which was absent in db/db OBESITY STUDIES fi term nutrition status as well as acute energy balance. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Technology for Proteomics Translation to Clinical Research
TechnologyTechnology ForFor ProteomicsProteomics Translation to Clinical ResearchResearch StudiesStudies Lance A. Liotta MD PhD George Mason University A. Novel one step preservative for tissue phosphoproteins B. Protein Microarrays: 200 signal pathway phosphoproteins -Translation to clinical research trials -The universal tissue preservative: obviate frozen storage C. Smart nanoparticles for one step in-solution molecular size sieving, affinity capture, biomarker preservation and amplification of effective sensitivity. The Center for Applied Proteomics and M ole cular Medicine Proteomics Tools for Clinical Medicine There is a need to measure the state of activity of the actual drug targets (the proteins) in a patient’s individual cancer. Patient A Patient B Proteomics is the missing link for designing individualized therapies Concurrent phosphorylation of kinases and kinase substrates indicates functional linkage “Proteins carry the epigenetic marks and information” Emma Whitelaw, DISCOVER Nov. 2006 Genetic or epigenetic defects are selected during cancer progression because they cooperate to orchestrate alterations in protein networks generating a survival advantage for the target cell. Post-translational modifications, such as phosphorylation, reflect the activity state of cellular signaling networks. Patterns of phosphorylation indicate docking events and infer protein-protein interactions. Pre‐analytical Variables: The tissue is alive! •The tissue is alive and reactive post excision • During the post excision delay time, tissue signal -

Chromatographic Studies of Protein-Based Chiral Separations
separations Review Chromatographic Studies of Protein-Based Chiral Separations Cong Bi, Xiwei Zheng, Shiden Azaria, Sandya Beeram, Zhao Li and David S. Hage * Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588-0304, USA; [email protected] (C.B.); [email protected] (X.Z.); [email protected] (S.A.); [email protected] (S.B.); [email protected] (Z.L.) * Correspondence: [email protected]; Tel.: +1-402-472-2744; Fax: +1-402-472-9402 Academic Editor: W John Lough Received: 12 June 2016; Accepted: 12 August 2016; Published: 5 September 2016 Abstract: The development of separation methods for the analysis and resolution of chiral drugs and solutes has been an area of ongoing interest in pharmaceutical research. The use of proteins as chiral binding agents in high-performance liquid chromatography (HPLC) has been an approach that has received particular attention in such work. This report provides an overview of proteins that have been used as binding agents to create chiral stationary phases (CSPs) and in the use of chromatographic methods to study these materials and protein-based chiral separations. The supports and methods that have been employed to prepare protein-based CSPs will also be discussed and compared. Specific types of CSPs that are considered include those that employ serum transport proteins (e.g., human serum albumin, bovine serum albumin, and alpha1-acid glycoprotein), enzymes (e.g., penicillin G acylase, cellobiohydrolases, and α-chymotrypsin) or other types of proteins (e.g., ovomucoid, antibodies, and avidin or streptavidin). The properties and applications for each type of protein and CSP will also be discussed in terms of their use in chromatography and chiral separations. -

Orosomucoid, a New Biomarker in the Association Between Obesity and Periodontitis
Orosomucoid, a new biomarker in the association between obesity and periodontitis. Hélène Rangé, Christine Poitou, Adrien Boillot, Cécile Ciangura, Sandrine Katsahian, Jean-Marc Lacorte, Sébastien Czernichow, Olivier Meilhac, Philippe Bouchard, Catherine Chaussain To cite this version: Hélène Rangé, Christine Poitou, Adrien Boillot, Cécile Ciangura, Sandrine Katsahian, et al.. Oroso- mucoid, a new biomarker in the association between obesity and periodontitis.. PLoS ONE, Public Library of Science, 2013, 8 (3), pp.e57645. 10.1371/journal.pone.0057645. hal-01446905 HAL Id: hal-01446905 https://hal.archives-ouvertes.fr/hal-01446905 Submitted on 26 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Orosomucoid, a New Biomarker in the Association between Obesity and Periodontitis He´le`ne Range´ 1,2, Christine Poitou3,4,5,6,7, Adrien Boillot1,8,Ce´cile Ciangura3,4,5, Sandrine Katsahian9, Jean-Marc Lacorte10,Se´bastien Czernichow8,11, Olivier Meilhac2, Philippe Bouchard1*, Catherine Chaussain12 1 Department of Periodontology, Service of Odontology, -
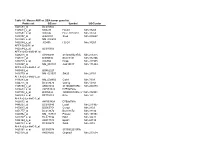
Table S1: Mouse AKR Vs DBA Tumor Gene List Probe Set GB Acc Symbol
Table S1: Mouse AKR vs DBA tumor gene list Probe set GB acc Symbol UGCluster 1425583_at BC010605 1425614_x_at M83244 H2-D1 Mm.33263 1427651_x_at X00246 H2-L /// H2-D1 Mm.33263 1419157_at AI428101 Sox4 Mm.240627 1422565_s_at NM_008688 1452544_x_at J00406 H2-D1 Mm.33263 AFFX-BioB-M_at 1425584_x_at BC010605 AFFX-r2-Ec-bioB-M_at 1426278_at AY090098 2310061N23Rik Mm.271275 1423411_at BI099836 BC013481 Mm.332406 1459725_s_at C86550 Dcpp Mm.287985 1419327_at NM_053181 AA415817 Mm.171484 AFFX-r2-Ec-bioB-3_at 1455869_at BG862223 1416770_at NM_021537 Stk25 Mm.28761 AFFX-r2-Ec-bioB-5_at 1418283_at NM_009903 Cldn4 Mm.7339 1424775_at BC018470 Oas1g Mm.14301 1428850_x_at AK004342 2410026K10Rik Mm.260878 1426633_s_at AW553424 D7Ertd760e 1427932_s_at BI076834 1200003I10Rik /// 1200015M12RikMm.332931 /// 1200016E24Rik 1449289_a_at BF715219 B2m Mm.163 AFFX-r2-Ec-bioC-3_at 1426632_at AW553424 D7Ertd760e 1448207_at BC010840 Lasp1 Mm.271967 1450017_at BG065754 Ccng1 Mm.2103 1451777_at BC013672 BC013672 Mm.33332 1420352_at NM_133731 Prss22 Mm.157351 1423747_a_at BC027196 Pdk1 Mm.34411 1454169_a_at AK017174 Epsti1 Mm.68134 1448793_a_at BC005679 Sdc4 Mm.3815 AFFX-r2-Ec-bioC-5_at 1425161_a_at BC005574 5730502D15Rik 1423158_at AK008566 Gnpnat1 Mm.233534 1453196_a_at BQ033138 Oasl2 Mm.228363 1449250_at NM_033573 Prcc Mm.35089 1452428_a_at AI099111 B2m Mm.163 1421024_at BB524140 Agpat1 Mm.8684 1450016_at BG065754 Ccng1 Mm.2103 1419043_a_at BM239828 AW111922 Mm.326506 1449262_s_at BB704337 Lin7c Mm.235300 1426975_at BG067859 4632413K17Rik Mm.295246 1426164_a_at AF479773 -

Urinary Proteomics and the Role of Orosomucoid (ORM) in Vascularization of Bladder Cancer
Urinary proteomics and the role of orosomucoid (ORM) in vascularization of bladder cancer Inaugural-Dissertation Zur Erlangung des Doktorgrads der Naturwissenschaften (Dr. rer. nat.) am Fachbereich Chemie an der Universität Duisburg-Essen vorgelegt von Ster Irmak aus Mardin Essen 2007 Die der vorliegenden Arbeit zugrunde liegenden Experimente wurden am Institut für Anatomie der Universität Duisburg-Essen und des Universitätsklinikums Hamburg-Eppendorf durchgeführt. 1. Gutachter: Prof. Dr. R. Sustmann 2. Gutachter: Prof. Dr. Dr. H. de Groot 3. Gutachter: Prof. Dr. S. Ergün Vorsitzender des Prüfungsausschusses: Prof. Dr. A. Schönbucher Tag der mündlichen Prüfung: 03.07.2007 To my Family and nephew Yusuf Heja Ever tried, ever failed. Try again, fail again. Fail better… Samuel Beckett Acknowledgement Acknowledgements The present study has been carried out between March 2003 - August 2006 at the Department of Anatomy, University Hospital Hamburg-Eppendorf, Germany and between August 2006 – March 2007 at the Department of Anatomy, University Hospital Essen, Germany. I wish to express my sincere gratitude to everyone who aided, supported and inspired me, in one way to another, throughout this study. First of all, I would especially like to thank my supervisor Professor Dr. Süleyman Ergün for introducing me to work in an exciting field of research, for his constant interest and support in the progress of the study and for the friendly atmosphere within the department. From the Department of Urology, University Hospital Hamburg-Eppendorf, I would like to thank Professor Dr. Huland for his support and the opportunity to work in the laboratory of the Urological Department. I would specially like to thank PD Dr. -

Indoxyl Sulfate and P-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance
BASIC RESEARCH www.jasn.org Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance Britt Opdebeeck ,1 Stuart Maudsley,2,3 Abdelkrim Azmi,3 Annelies De Maré,1 Wout De Leger,4 Bjorn Meijers,5,6 Anja Verhulst,1 Pieter Evenepoel,5,6 Patrick C. D’Haese,1 and Ellen Neven1 1Laboratory of Pathophysiology, Department of Biomedical Sciences, 2Receptor Biology Lab, Department of Biomedical Sciences, and 3Translational Neurobiology Group, Flanders Institute of Biotechnology Center for Molecular Neurology, Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium; 4Division of Molecular Design and Synthesis, Department of Chemistry and 6Laboratory of Nephrology, Department of Immunology and Microbiology, Catholic University of Leuven, Leuven, Belgium; and 5Division of Internal Medicine, Nephrology, University Hospitals Leuven, Leuven, Belgium ABSTRACT Background Protein-bound uremic toxins indoxyl sulfate (IS) and p-cresyl sulfate (PCS) have been associ- ated with cardiovascular morbidity and mortality in patients with CKD. However, direct evidence for a role of these toxins in CKD-related vascular calcification has not been reported. Methods To study early and late vascular alterations by toxin exposure, we exposed CKD rats to vehicle, IS (150 mg/kg per day), or PCS (150 mg/kg per day) for either 4 days (short-term exposure) or 7 weeks (long-term exposure). We also performed unbiased proteomic analyses of arterial samples coupled to functional bioinformatic annotation analyses to investigate molecular signaling events associated with toxin-mediated arterial calcification. Results Long-term exposure to either toxin at serum levels similar to those experienced by patients with CKD significantly increased calcification in the aorta and peripheral arteries.