P-00814 (12/2020) 4
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Case Study of Distinctive Phenotypes Arising from Emanuel Syndrome in Two Karyotypically Identical Patients Mot Yee Yik1, Rabiatul Basria S.M.N
Malaysian Journal of Medicine and Health Sciences (eISSN 2636-9346) CASE REPORT A Case Study of Distinctive Phenotypes Arising From Emanuel Syndrome in Two Karyotypically Identical Patients Mot Yee Yik1, Rabiatul Basria S.M.N. Mydin2, Emmanuel Jairaj Moses1, Shahrul Hafiz Mohd Zaini2,3, Abdul Rahman Azhari4, Narazah Mohd Yusoff1 1 Regenerative Medicine Sciences Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, 13200 Bertam, Kepala Batas, Pulau Pinang Malaysia 2 Oncological and Radiological Sciences Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, 13200 Bertam, Kepala Batas, Pulau Pinang Malaysia, 3 Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur. 4 Human Genetics Unit, Advanced Diagnostic Laboratory, Advanced Medical and Dental Institute, Universiti Sains Malaysia ABSTRACT Emanuel syndrome, also referred to as supernumerary der(22) or t(11;22) syndrome, is a rare genomic syndrome. Patients are normally presented with multiple congenital anomalies and severe developmental disabilities. Affected newborns usually carry a derivative chromosome 22 inherited from either parent, which stems from a balanced translocation between chromosomes 11 and 22. Unfortunately, identification of Emanuel syndrome carriers is diffi- cult as balanced translocations do not typically present symptoms. We identified two patients diagnosed as Emanuel syndrome with identical chromosomal aberration: 47,XX,+der(22)t(11;22)(q24;q12.1)mat karyotype but presenting variable phenotypic features. Emanuel syndrome patients present variable phenotypes and karyotypes have also been inconsistent albeit the existence of a derivative chromosome 22. Our data suggests that there may exist ac- companying genetic aberrations which influence the outcome of Emanuel syndrome phenotypes but it should be cautioned that more patient observations, diagnostic data and research is required before conclusions can be drawn on definitive karyotypic-phenotypic correlations. -
Disability Classification System
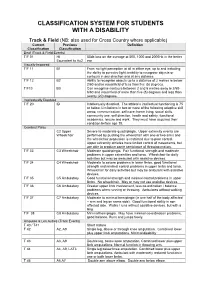
CLASSIFICATION SYSTEM FOR STUDENTS WITH A DISABILITY Track & Field (NB: also used for Cross Country where applicable) Current Previous Definition Classification Classification Deaf (Track & Field Events) T/F 01 HI 55db loss on the average at 500, 1000 and 2000Hz in the better Equivalent to Au2 ear Visually Impaired T/F 11 B1 From no light perception at all in either eye, up to and including the ability to perceive light; inability to recognise objects or contours in any direction and at any distance. T/F 12 B2 Ability to recognise objects up to a distance of 2 metres ie below 2/60 and/or visual field of less than five (5) degrees. T/F13 B3 Can recognise contours between 2 and 6 metres away ie 2/60- 6/60 and visual field of more than five (5) degrees and less than twenty (20) degrees. Intellectually Disabled T/F 20 ID Intellectually disabled. The athlete’s intellectual functioning is 75 or below. Limitations in two or more of the following adaptive skill areas; communication, self-care; home living, social skills, community use, self direction, health and safety, functional academics, leisure and work. They must have acquired their condition before age 18. Cerebral Palsy C2 Upper Severe to moderate quadriplegia. Upper extremity events are Wheelchair performed by pushing the wheelchair with one or two arms and the wheelchair propulsion is restricted due to poor control. Upper extremity athletes have limited control of movements, but are able to produce some semblance of throwing motion. T/F 33 C3 Wheelchair Moderate quadriplegia. Fair functional strength and moderate problems in upper extremities and torso. -

Reframing Psychiatry for Precision Medicine
Reframing Psychiatry for Precision Medicine Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

CADASIL Testing
Lab Management Guidelines V1.0.2020 CADASIL Testing MOL.TS.144.A v1.0.2020 Introduction CADASIL testing is addressed by this guideline. Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline NOTCH3 Known Familial Mutation 81403 Analysis NOTCH3 Targeted Sequencing 81406 NOTCH3 Deletion/Duplication Analysis 81479 What is CADASIL Definition CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) is an adult-onset form of cerebrovascular disease. There are no generally accepted clinical diagnostic criteria for CADASIL and symptoms vary among affected individuals. Signs and symptoms Typical signs and symptoms include1,2,3 Transient ischemic attacks and ischemic stroke, occurs at a mean age of 47 years (age range 20-70 years), in most cases without conventional vascular risk factors cognitive disturbance, primarily affecting executive function, may start as early as age 35 years psychiatric or behavioral abnormalities migraine with aura, occurs with a mean age of onset of 30 years (age range 6-48 years), and Less common symptoms include: © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V1.0.2020 recurrent seizures with onset in middle age, usually secondary to stroke acute encephalopathy, with a mean age of onset of 42 years Life expectancy for men with CADASIL is reduced by approximately five years and for women by 1 to 2 years.4 Diagnosis Brain Magnetic Resonance Imaging (MRI) findings include T2-signal-abnormalities in the white matter of the temporal pole and T2-signal-abnormalities in the external capsule and corpus callosum.1,2 CADASIL is suspected in an individual with the clinical signs and MRI findings. -

The National Economic Burden of Rare Disease Study February 2021
Acknowledgements This study was sponsored by the EveryLife Foundation for Rare Diseases and made possible through the collaborative efforts of the national rare disease community and key stakeholders. The EveryLife Foundation thanks all those who shared their expertise and insights to provide invaluable input to the study including: the Lewin Group, the EveryLife Community Congress membership, the Technical Advisory Group for this study, leadership from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH), the Undiagnosed Diseases Network (UDN), the Little Hercules Foundation, the Rare Disease Legislative Advocates (RDLA) Advisory Committee, SmithSolve, and our study funders. Most especially, we thank the members of our rare disease patient and caregiver community who participated in this effort and have helped to transform their lived experience into quantifiable data. LEWIN GROUP PROJECT STAFF Grace Yang, MPA, MA, Vice President Inna Cintina, PhD, Senior Consultant Matt Zhou, BS, Research Consultant Daniel Emont, MPH, Research Consultant Janice Lin, BS, Consultant Samuel Kallman, BA, BS, Research Consultant EVERYLIFE FOUNDATION PROJECT STAFF Annie Kennedy, BS, Chief of Policy and Advocacy Julia Jenkins, BA, Executive Director Jamie Sullivan, MPH, Director of Policy TECHNICAL ADVISORY GROUP Annie Kennedy, BS, Chief of Policy & Advocacy, EveryLife Foundation for Rare Diseases Anne Pariser, MD, Director, Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health Elisabeth M. Oehrlein, PhD, MS, Senior Director, Research and Programs, National Health Council Christina Hartman, Senior Director of Advocacy, The Assistance Fund Kathleen Stratton, National Academies of Science, Engineering and Medicine (NASEM) Steve Silvestri, Director, Government Affairs, Neurocrine Biosciences Inc. -

2020 Question Book
2020 QUESTION BOOK 13TH EDITION WHO WE ARE Welcome to the thirteenth edition of the Ninja’s Guide to PRITE! Loma Linda University Medical Center is located in sunny Southern California. about 60 miles east of Los Angeles. A part of the Adventist Health System, we provide patient care in one of the largest non-profit health systems in the nation. Loma Linda's mission is to excel in medical education, global healthcare, and community outreach, all under a central tenant: "To Make Man Whole." At the Loma Linda Department of Psychiatry, our residents are trained in many diverse patient care settings. As an official World Health Organization Collaboration Center, our department funds resident electives in Global Mental Health at locations around the world. Additionally, our residents can participate in national and international disaster relief on the LLU Behavioral Health Trauma Team. We were proud to welcome our first group of Child and Adolescent Psychiatry fellows in the Summer of 2019 and work collaboratively with 3 other residency programs within the region. Our residency didactic education is constantly evolving based upon resident feedback, and our residents have the opportunity to aid in course development. More than anything, our residency fosters an environment where residents and faculty treat each other like family. Our faculty are dedicated to resident education and professional development. We believe in "taking 'No' off the table", encouraging innovative change, and passionately supporting our residents to achieve anything they set their minds to. For over a decade our residents have volunteered their time to create The Ninja's Guide to PRITE at our Annual Ninja PRITE Workshop. -

Ifds Functional Classification System & Procedures
IFDS FUNCTIONAL CLASSIFICATION SYSTEM & PROCEDURES MANUAL 2009 - 2012 Effective – 1 January 2009 Originally Published – March 2009 IFDS, C/o ISAF UK Ltd, Ariadne House, Town Quay, Southampton, Hampshire, SO14 2AQ, GREAT BRITAIN Tel. +44 2380 635111 Fax. +44 2380 635789 Email: [email protected] Web: www.sailing.org/disabled 1 Contents Page Introduction 5 Part A – Functional Classification System Rules for Sailors A1 General Overview and Sailor Evaluation 6 A1.1 Purpose 6 A1.2 Sailing Functions 6 A1.3 Ranking of Functional Limitations 6 A1.4 Eligibility for Competition 6 A1.5 Minimum Disability 7 A2 IFDS Class and Status 8 A2.1 Class 8 A2.2 Class Status 8 A2.3 Master List 10 A3 Classification Procedure 10 A3.0 Classification Administration Fee 10 A3.1 Personal Assistive Devices 10 A3.2 Medical Documentation 11 A3.3 Sailors’ Responsibility for Classification Evaluation 11 A3.4 Sailor Presentation for Classification Evaluation 12 A3.5 Method of Assessment 12 A3.6 Deciding the Class 14 A4 Failure to attend/Non Co-operation/Misrepresentation 16 A4.1 Sailor Failure to Attend Evaluation 16 A4.2 Non Co-operation during Evaluation 16 A4.3 International Misrepresentation of Skills and/or Abilities 17 A4.4 Consequences for Sailor Support Personnel 18 A4.5 Consequences for Teams 18 A5 Specific Rules for Boat Classes 18 A5.1 Paralympic Boat Classes 18 A5.2 Non-Paralympic Boat Classes 19 Part B – Protest and Appeals B1 Protest 20 B1.1 General Principles 20 B1.2 Class Status and Protest Opportunities 21 B1.3 Parties who may submit a Classification Protest -

Consensus Statement on Immune Modulation in Multiple Sclerosis
[Downloaded free from http://www.annalsofian.org on Monday, June 8, 2020, IP: 202.164.42.67] View Point Consensus Statement On Immune Modulation in Multiple Sclerosis and Related Disorders During the COVID‑19 Pandemic: Expert Group on Behalf of the Indian Academy of Neurology Rohit Bhatia, M. V. Padma Srivastava, Dheeraj Khurana1, Lekha Pandit2, Thomas Mathew3, Salil Gupta4, Netravathi M5, Sruthi S. Nair6, Gagandeep Singh7, Bhim S. Singhal8 Department of Neurology, All India Institute of Medical Sciences, New Delhi, 1Department of Neurology, Postgraduate Institute of Medical Education and Research, Chandigarh, 2Department of Neurology, K.S. Hegde Medical Academy, Mangalore, Karnataka, 3Department of Neurology, St John’s Medical College, Bangalore, Karnataka, 4Department of Neurology, Command Hospital Air Force, Bangalore, Karnataka, 5Department of Neurology, National Institute of Mental Health and Neurosciences, Bangalore, Karnataka, 6Department of Neurology, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Thiruvananthapuram, Kerala, 7Department of Neurology, Dayanand Medical College and Hospital, Ludhiana, Punjab, 8Department of Neurology, Bombay Hospital, Mumbai, Maharashtra, India Abstract Knowledge related to SARS-CoV-2 or 2019 novel coronavirus (2019-nCoV) is still emerging and rapidly evolving. We know little about the effects of this novel coronavirus on various body systems and its behaviour among patients with underlying neurological conditions, especially those on immunomodulatory medications. The aim of the present consensus expert opinion document is to appraise the potential concerns when managing our patients with underlying CNS autoimmune demyelinating disorders during the current COVID-19 pandemic. Keywords: COVID 19, disease modifying therapy, immunotherapy, MOG, MS, NMOSD INTRODUCTION cough, myalgia and headache with acute respiratory distress syndrome (ARDS) being the most frequent complication, In December 2019, health authorities recognized an outbreak inducing mortality in six (15%) patients. -

ICD-9CM Coding Achilles Bursitis Or Tendinitis 726.71 Adhesive
ICD-9CM CODING OF COMMON CHIROPRACTIC CONDITIONS CONDITION ICD-9CM coding Achilles bursitis or tendinitis 726.71 Adhesive capsulitis of shoulder 726 Anklyosing Spondylitis (spine only) 720 Anterior cruciate ligament (old disruption) 717.83 Bicipital tenosynovitis 726.12 Boutonniere deformity 736.21 Brachial neuritis or radiculitis 723.4 Bunion 727.1 Bursitis of knee 726.6 Calcaneal spur 726.73 Carpal tunnel syndrome 354 Cervical radiculitis 723.4 Cervical spondylosis with myelopathy 721.1 Cervical spondylosis without myelopathy 721 Cervicalgia 723.1 Chondromalacia of patella 717.7 Claw hand (acquired) 736.06 Claw toe (acquired) 735.5 Coccygodynia 724.79 Coxa plana 732.1 Coxa valga (acquired) 736.31 Coxa vara (acquired) 736.32 de Quervain's disease 727.04 Degeneration of cervical intervertebral disc 722.4 Degeneration of thoracic or lumbar intervertebral disc 722.5 Disc Degeneration (Cervical with myelopathy) 722.71 Disc Degeneration (Lumbar with myelopathy) 722.73 Disc Degeneration (Thoracic) 722.51 Disc Degeneration (Cervical) 722.4 Disc Degeneration (Lumbar) 722.52 Disc Degeneration (Thoracic with myelopathy) 722.72 Disc displacement without myelopathy (Thoracic) 722.11 Disc displacement of without myelopathy (Cervical ) 722 Disc displacement without myelopathy (Lumbar) 722.1 Dupuytren's contracture 728.6 Entrapment syndromes 354.0-355.9 Epicondylitis (Lateral) 726.32 Epicondylitis (Medial) 726.31 Flat foot-Pes planus (acquired) 734 Fracture (Lumbar) 805.4 Ganglion of tendon sheath 727.42 Genu recurvatum (acquired) 736.5 Genu valgum -

Malignant Hyperthermia
:: Malignant hyperthermia Synonyms: malignant hyperpyrexia, hyperthermia of anesthesia Syndromes with higher risk of MH: ` King-Denborough syndrome ` central core disease (CCD, central core myopathy) ` multiminicore disease (with or without RYR1 mutation) ` nemaline rod myopathy (with or without RYR1 mutation) ` hypokalemic periodic paralysis Definition: Malignant hyperthermia (MH) is a rare disorder of skeletal muscles related to a high release of calcium from the sarcoplasmic reticulum which leads to muscle rigidity in many cases and hypermetabolism. Abrupt onset is triggered either by anesthesic agents such as halogenated volatile anesthetics and depolarizing muscle relaxant such as succinylcholine (MH of anesthesia), or, occasionally, by stresses such as vigorous exercise or heat. In most cases, mutations of RYR1 and CACNA1S genes have been reported. MH is characterized by tachycardia, arrhythmia, muscle rigidity, hyperthermia, skin mottling, rhabdomyolysis (cola- colored urine) metabolic acidosis, electrolyte disturbances especially hyperkalemia and coagulopathy. Dantrolene is currently the only known treatment for a MH crisis. Further information: See the Orphanet abstract Menu Pre-hospital emergency care Recommendations for hospital recommendations emergency departments Synonyms Emergency issues Aetiology Emergency recommendations Special risks in emergency situations Management approach Frequently used long term treatments Drug interactions Complications Anesthesia Specific medical care prior to hospitalisation Preventive measures -

A Case Report on Charcot-Marie-Tooth Disease with a Novel Periaxin Gene Mutation
Open Access Case Report DOI: 10.7759/cureus.5111 A Case Report on Charcot-Marie-Tooth Disease with a Novel Periaxin Gene Mutation Sorabh Datta 1 , Saurabh Kataria 1 , Raghav Govindarajan 1 1. Neurology, University of Missouri, Columbia, USA Corresponding author: Sorabh Datta, [email protected] Abstract Charcot-Marie-Tooth (CMT) disease is one of the most common primary hereditary neuropathies causing peripheral neuropathies. More than 60 different gene mutations are causing this disease. The PRX gene codes for Periaxin proteins that are expressed by Schwann cells and are necessary for the formation and maintenance of myelination of peripheral nerves. Dejerine-Sottas neuropathy and Charcot-Marie-Tooth type 4F (CMT4F) are the two different clinical phenotypes observed in association with PRX gene mutation. This article describes a case of an elderly male with a novel mutation involving the PRX gene. Categories: Genetics, Internal Medicine, Neurology Keywords: neurology, sensorimotor neuropathy, congenital, gene expression, genetic mutation, protein, pes cavus, demyelinating diseases, charcot-marie-tooth, autosomal recessive disorder Introduction As per the Dyck classification in the year 1970, primary hereditary neuropathies are divided into hereditary motor sensory neuropathy (HMSN) and hereditary sensory autonomic neuropathy (HSAN) [1]. Charcot- Marie-Tooth (CMT) disease is a type of HMSN with an estimated prevalence of 1 in 2,500 [2]. CMT can follow autosomal recessive (ARCMT), X-linked recessive, and also an autosomal dominant pattern. CMT type 4 is a rapidly increasing ARCMT disease form in HMSN, although CMT type 1 and 2 still account for the most substantial proportion of the patient population [3]. CMT4F is a severe, demyelinating subtype of CMT type 4 and is characterized by childhood onset of slowly progressing weakness in the distal muscles associated with atrophy. -

Sensory Deficits in a Mouse Model of Christianson Syndrome Tarheen
Sensory Deficits in a Mouse Model of Christianson Syndrome Tarheen Fatima Department of Physiology McGill University, Montreal April 2019 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Master of Science. © Tarheen Fatima 2019 Abstract Christianson Syndrome (CS) is a recently characterized X-linked neurodevelopmental disorder caused by loss-of-function mutations in the gene slc9a6, encoding the endosomal Na+/H+ Exchanger 6 (NHE6). The disorder is associated with developmental delay, intellectual disability, loss of motor coordination, mutism, ataxia, epilepsy, as well as autistic features. In addition to these symptoms, CS patients exhibit elevated pain thresholds to noxious stimuli as well as discomfort at normally innocuous stimuli. The underlying causes of these sensory deficits are yet to be determined. Hence, this study aims at understanding how loss-of-function of NHE6 affects transmission and processing of pain. To this end, we examined the expression of NHE6 in peripheral and central neurons implicated in sensation and interpretation of pain. Additionally, we characterized the nociceptive behaviour of NHE6 knockout (KO) mice using a battery of behaviour tests. Our immunohistochemical experiments demonstrate that NHE6 is highly expressed in nociceptive, small-diameter dorsal root ganglia (DRG) neurons. Moreover, mice lacking NHE6 display decreased responses to noxious mechanical, thermal and chemical stimuli but are more responsive to noxious cold than wild-type littermates. Interestingly, immunohistochemical characterization of DRG tissue from aged NHE6 null mice indicates a decrease in some neuronal subsets suggesting cell death. Finally, using light brush-induced Fos activation in the dorsal horn, we found that the spinal processing of innocuous stimuli is not different between wildtype and NHE6 knockout mice.