Ribosome Assembly, Proteolysis and Pathogenesis in Human Mitochondria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Proteomic Analysis of the Role of the Quality Control Protease LONP1 in Mitochondrial Protein Aggregation
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.12.439502; this version posted April 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Proteomic analysis of the role of the quality control protease LONP1 in mitochondrial protein aggregation Karen Pollecker1, Marc Sylvester2 and Wolfgang Voos1,* 1Institute of Biochemistry and Molecular Biology (IBMB), University of Bonn, Faculty of Medicine, Nussallee 11, 53115 Bonn, Germany 2Core facility for mass spectrometry, Institute of Biochemistry and Molecular Biology (IBMB), University of Bonn, Faculty of Medicine, Nussallee 11, 53115 Bonn, Germany *Corresponding author Email: [email protected] Phone: +49-228-732426 Abbreviations: AAA+, ATPases associated with a wide variety of cellular activities; Δψ, mitochondrial membrane potential; gKD, genetic knockdown; HSP, heat shock protein; m, mature form; mt, mitochondrial; p, precursor form; PQC, protein quality control; qMS, quantitative mass spectrometry; ROS, reactive oxygen species; SILAC, stable isotope labeling with amino acids in cell culture; siRNA, small interfering RNA; TIM, preprotein translocase complex of the inner membrane; TMRE, tetramethylrhodamine; TOM, preprotein translocase complex of the outer membrane; UPRmt, mitochondrial unfolded protein response; WT, wild type. bioRxiv preprint doi: https://doi.org/10.1101/2021.04.12.439502; this version posted April 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Reprogramming of Trna Modifications Controls the Oxidative Stress Response by Codon-Biased Translation of Proteins
Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Chan, Clement T.Y. et al. “Reprogramming of tRNA Modifications Controls the Oxidative Stress Response by Codon-biased Translation of Proteins.” Nature Communications 3 (2012): 937. As Published http://dx.doi.org/10.1038/ncomms1938 Publisher Nature Publishing Group Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/76775 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins Clement T.Y. Chan,1,2 Yan Ling Joy Pang,1 Wenjun Deng,1 I. Ramesh Babu,1 Madhu Dyavaiah,3 Thomas J. Begley3 and Peter C. Dedon1,4* 1Department of Biological Engineering, 2Department of Chemistry and 4Center for Environmental Health Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139; 3College of Nanoscale Science and Engineering, University at Albany, SUNY, Albany, NY 12203 * Corresponding author: PCD, Department of Biological Engineering, NE47-277, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139; tel 617-253-8017; fax 617-324-7554; email [email protected] 2 ABSTRACT Selective translation of survival proteins is an important facet of cellular stress response. We recently demonstrated that this translational control involves a stress-specific reprogramming of modified ribonucleosides in tRNA. -

Dissert 1 Title
GENETIC AND BIOCHEMICAL ANALYSIS OF ESSENTIAL ENZYMES IN TRIACYLGLYCEROL SYNTHESIS IN ARABIDOPSIS By KIMBERLY LYNN COTTON A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY WASHINGTON STATE UNIVERSITY Molecular Plant Sciences DECEMBER 2015 © Copyright by KIMBERLY LYNN COTTON, 2015 All Rights Reserved To the Faculty of Washington State University: The members of the Committee appointed to examine the dissertation of KIMBERLY LYNN COTTON find it satisfactory and recommend that it be accepted. ____________________________________________ John A. Browse, Ph.D., Chair _____________________________________________ Michael M. Neff, Ph.D. _____________________________________________ Thomas W. Okita, Ph.D. _____________________________________________ John J. Wyrick, Ph.D. ii ACKNOWLEDGMENTS To my PI John Browse, thank you for your support, guidance and patience over the years. To my committee, John Browse, Michael Neff, Thomas Okita, and John Wyrick, thank you for your insightful discussions and suggestions. To my collaborators Jay Shockey, Anushobha Regmi, Neil Adhikari, John Browse, and Phil Bates, thanks for the great work and great times. To Browse Lab members past and present, thanks for the help, guidance and wonderful memories. To the undergraduate students who have helped me over the years, thank you for all your help in making this project possible; and with a special thank you to Arlene, Tesa, Breana, and “no-longer-undergrads” David and Barbara Cotton for their particular dedication to this project. And finally, a huge thank you to my family and friends who have loved and supported me throughout this venture. Go Cougs! iii GENETIC AND BIOCHEMICAL ANALYSIS OF ESSENTIAL ENZYMES IN TRIACYLGLYCEROL SYNTHESIS IN ARABIDOPSIS Abstract by Kimberly Lynn Cotton, Ph.D. -

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Cytotoxic Effects and Changes in Gene Expression Profile
Toxicology in Vitro 34 (2016) 309–320 Contents lists available at ScienceDirect Toxicology in Vitro journal homepage: www.elsevier.com/locate/toxinvit Fusarium mycotoxin enniatin B: Cytotoxic effects and changes in gene expression profile Martina Jonsson a,⁎,MarikaJestoib, Minna Anthoni a, Annikki Welling a, Iida Loivamaa a, Ville Hallikainen c, Matti Kankainen d, Erik Lysøe e, Pertti Koivisto a, Kimmo Peltonen a,f a Chemistry and Toxicology Research Unit, Finnish Food Safety Authority (Evira), Mustialankatu 3, FI-00790 Helsinki, Finland b Product Safety Unit, Finnish Food Safety Authority (Evira), Mustialankatu 3, FI-00790 Helsinki, c The Finnish Forest Research Institute, Rovaniemi Unit, P.O. Box 16, FI-96301 Rovaniemi, Finland d Institute for Molecular Medicine Finland (FIMM), University of Helsinki, P.O. Box 20, FI-00014, Finland e Plant Health and Biotechnology, Norwegian Institute of Bioeconomy, Høyskoleveien 7, NO -1430 Ås, Norway f Finnish Safety and Chemicals Agency (Tukes), Opastinsilta 12 B, FI-00521 Helsinki, Finland article info abstract Article history: The mycotoxin enniatin B, a cyclic hexadepsipeptide produced by the plant pathogen Fusarium,isprevalentin Received 3 December 2015 grains and grain-based products in different geographical areas. Although enniatins have not been associated Received in revised form 5 April 2016 with toxic outbreaks, they have caused toxicity in vitro in several cell lines. In this study, the cytotoxic effects Accepted 28 April 2016 of enniatin B were assessed in relation to cellular energy metabolism, cell proliferation, and the induction of ap- Available online 6 May 2016 optosis in Balb 3T3 and HepG2 cells. The mechanism of toxicity was examined by means of whole genome ex- fi Keywords: pression pro ling of exposed rat primary hepatocytes. -

Construction of Prognostic Microrna Signature for Human Invasive Breast Cancer by Integrated Analysis
Journal name: OncoTargets and Therapy Article Designation: Original Research Year: 2019 Volume: 12 OncoTargets and Therapy Dovepress Running head verso: Shi et al Running head recto: Shi et al open access to scientific and medical research DOI: 189265 Open Access Full Text Article ORIGINAL RESEARCH Construction of prognostic microRNA signature for human invasive breast cancer by integrated analysis This article was published in the following Dove Medical Press journal: OncoTargets and Therapy Wei Shi* Background: Despite the advances in early detection and treatment methods, breast cancer Fang Dong* still has a high mortality rate, even in those patients predicted to have a good prognosis. The Yujia Jiang purpose of this study is to identify a microRNA signature that could better predict prognosis in Linlin Lu breast cancer and add new insights to the current classification criteria. Changwen Wang Materials and methods: We downloaded microRNA sequencing data along with correspond- Jie Tan ing clinicopathological data from The Cancer Genome Atlas (TCGA). Of 1,098 breast cancer patients identified, 253 patients with fully characterized microRNA profiles were selected for Wen Yang analysis. A three-microRNA signature was generated in the training set. Subsequently, the per- Hui Guo formance of the signature was confirmed in a validation set. After construction of the signature, Jie Ming* we conducted additional experiments, including flow cytometry and the Cell Counting Kit-8 Tao Huang* assay, to illustrate the correlation of this microRNA signature with breast cancer cell cycle, Department of Breast and Thyroid apoptosis, and proliferation. Surgery, Union Hospital, Tongji Results: Three microRNAs (hsa-mir-31, hsa-mir-16-2, and hsa-mir-484) were identified to Medical College, Huazhong University of Science and Technology, Wuhan be significantly and independently correlated with patient prognosis, and performed with good 430022, China stability. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Anti-Inflammatory Role of Curcumin in LPS Treated A549 Cells at Global Proteome Level and on Mycobacterial Infection
Anti-inflammatory Role of Curcumin in LPS Treated A549 cells at Global Proteome level and on Mycobacterial infection. Suchita Singh1,+, Rakesh Arya2,3,+, Rhishikesh R Bargaje1, Mrinal Kumar Das2,4, Subia Akram2, Hossain Md. Faruquee2,5, Rajendra Kumar Behera3, Ranjan Kumar Nanda2,*, Anurag Agrawal1 1Center of Excellence for Translational Research in Asthma and Lung Disease, CSIR- Institute of Genomics and Integrative Biology, New Delhi, 110025, India. 2Translational Health Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, 110067, India. 3School of Life Sciences, Sambalpur University, Jyoti Vihar, Sambalpur, Orissa, 768019, India. 4Department of Respiratory Sciences, #211, Maurice Shock Building, University of Leicester, LE1 9HN 5Department of Biotechnology and Genetic Engineering, Islamic University, Kushtia- 7003, Bangladesh. +Contributed equally for this work. S-1 70 G1 S 60 G2/M 50 40 30 % of cells 20 10 0 CURI LPSI LPSCUR Figure S1: Effect of curcumin and/or LPS treatment on A549 cell viability A549 cells were treated with curcumin (10 µM) and/or LPS or 1 µg/ml for the indicated times and after fixation were stained with propidium iodide and Annexin V-FITC. The DNA contents were determined by flow cytometry to calculate percentage of cells present in each phase of the cell cycle (G1, S and G2/M) using Flowing analysis software. S-2 Figure S2: Total proteins identified in all the three experiments and their distribution betwee curcumin and/or LPS treated conditions. The proteins showing differential expressions (log2 fold change≥2) in these experiments were presented in the venn diagram and certain number of proteins are common in all three experiments. -
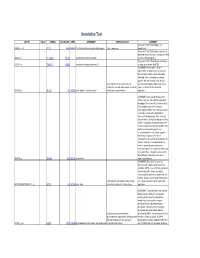
Supplementary Table 5. Functional Annotation of the Largest Gene Cluster(221 Element)
Annotation Tool AFFYID VALUE SYMBOL LOCUSLINK OMIM GENENAME GENEONTOLOGY SUMMARY [Proteome FUNCTION:] Expressed 203054_s_at TCTA 6988 600690 T-cell leukemia translocation altered gene tumor suppressor ubiquitously [Proteome FUNCTION:] May be involved in protein-protein interactions; contains five WD 44563_at FLJ10385 55135 hypothetical protein FLJ10385 domains (WD-40 repeats) [Proteome FUNCTION:] Weakly similarity to 212261_at TNRC15 26058 trinucleotide repeat containing 15 a region of rat nestin (Rn.9701) [SUMMARY:] Actin alpha 1 which is expressed in skeletal muscle is one of six different actin isoforms which have been identified. Actins are highly conserved proteins that are involved in cell motility, actin filament; motor activity; muscle structure and integrity. Alpha actins are a contraction; muscle development; structural major constituent of the contractile 203872_at ACTA1 58 102610 actin, alpha 1, skeletal muscle constituent of cytoskeleton apparatus. [SUMMARY:] Annexin VIII belong to the family of Ca (2+) dependent phospholipid binding proteins (annexins), and has a high 56% identity to annexin V (vascular anticoagulant-alpha). It was initially isolated as 2.2 kb vascular anticoagulant-beta transcript from human placenta, a Ca (2+) dependent phospholipid binding protein that inhibits coagulation and phospholipase A2 activity. However, the fact that annexin VIII is neither an extracellular protein nor associated with the cell surface suggests that it may not play a role in blood coagulation in vivo and its physiological role remains unknown. It is expressed at low levels in human placenta and shows restricted expression in lung endothelia, skin, liver, and kidney. The gene is also found to be selectively overexpressed in acute 203074_at ANXA8 244 602396 annexin A8 myelocytic leukemia. -

Structural Basis for Late Maturation Steps of the Human Mitoribosomal Large Subunit ✉ ✉ Miriam Cipullo1,2,5, Genís Valentín Gesé3,5, Anas Khawaja1,2, B
ARTICLE https://doi.org/10.1038/s41467-021-23617-8 OPEN Structural basis for late maturation steps of the human mitoribosomal large subunit ✉ ✉ Miriam Cipullo1,2,5, Genís Valentín Gesé3,5, Anas Khawaja1,2, B. Martin Hällberg 3,4 & Joanna Rorbach 1,2 Mitochondrial ribosomes (mitoribosomes) synthesize a critical set of proteins essential for oxidative phosphorylation. Therefore, mitoribosomal function is vital to the cellular energy supply. Mitoribosome biogenesis follows distinct molecular pathways that remain poorly 1234567890():,; understood. Here, we determine the cryo-EM structures of mitoribosomes isolated from human cell lines with either depleted or overexpressed mitoribosome assembly factor GTPBP5, allowing us to capture consecutive steps during mitoribosomal large subunit (mt- LSU) biogenesis. Our structures provide essential insights into the last steps of 16S rRNA folding, methylation and peptidyl transferase centre (PTC) completion, which require the coordinated action of nine assembly factors. We show that mammalian-specific MTERF4 contributes to the folding of 16S rRNA, allowing 16 S rRNA methylation by MRM2, while GTPBP5 and NSUN4 promote fine-tuning rRNA rearrangements leading to PTC formation. Moreover, our data reveal an unexpected involvement of the elongation factor mtEF-Tu in mt-LSU assembly, where mtEF-Tu interacts with GTPBP5, similar to its interaction with tRNA during translational elongation. 1 Department of Medical Biochemistry and Biophysics, Division of Molecular Metabolism, Karolinska Institutet, Solna, Sweden. 2 Max Planck Institute Biology of Ageing—Karolinska Institutet Laboratory, Karolinska Institutet, Stockholm, Sweden. 3 Department of Cell and Molecular Biology, Karolinska Institutet, Solna, Sweden. 4 Centre for Structural Systems Biology (CSSB) and Karolinska Institutet VR-RÅC, Hamburg, Germany. -

Mitochondrial Lon Protease in Human Disease and Aging: Including an Etiologic Classification of Lon-Related Diseases and Disorders
UC Irvine UC Irvine Previously Published Works Title Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Permalink https://escholarship.org/uc/item/2pw606qk Authors Bota, Daniela A Davies, Kelvin JA Publication Date 2016-11-01 DOI 10.1016/j.freeradbiomed.2016.06.031 Peer reviewed eScholarship.org Powered by the California Digital Library University of California HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Free Radic Manuscript Author Biol Med. Author Manuscript Author manuscript; available in PMC 2016 December 24. Published in final edited form as: Free Radic Biol Med. 2016 November ; 100: 188–198. doi:10.1016/j.freeradbiomed.2016.06.031. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders Daniela A. Botaa,* and Kelvin J.A. Daviesb,c a Department of Neurology and Chao Family Comprehensive Cancer Center, UC Irvine School of Medicine, 200 S. Manchester Ave., Suite 206, Orange, CA 92868, USA b Leonard Davis School of Gerontology of the Ethel Percy Andrus Gerontology Center, Los Angeles, CA 90089-0191, USA c Division of Molecular & Computational Biology, Department of Biological Sciences, Dornsife College of Letters, Arts, & Sciences, The University of Southern California, Los Angeles, CA 90089-0191, USA Abstract The Mitochondrial Lon protease, also called LonP1 is a product of the nuclear gene LONP1. Lon is a major regulator of mitochondrial metabolism and response to free radical damage, as well as an essential factor for the maintenance and repair of mitochondrial DNA. Lon is an ATP- stimulated protease that cycles between being bound (at the inner surface of the inner mitochondrial membrane) to the mitochondrial genome, and being released into the mitochondrial matrix where it can degrade matrix proteins.