Spinal Muscular Atrophy : Evidence of a Multi-System Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reports of Committees
PART II REPORTS OF COMMITTEES The text of resolutions and decisions recommended in the reports of Committee A and Committee B and subsequently adopted by the Health Assembly has been replaced by the respective resolution or decision number (in square brackets). The verbatim records of the plenary meetings at which these reports were approved are available on the WHO website, official records page: http://apps.who.int/gb/or/. COMMITTEE ON CREDENTIALS Report 1 [A66/64 – 21 May 2013] The Committee on Credentials met on 21 May 2013. Delegates of the following Member States were present: Canada; Cook Islands; Malawi; Mali; Mongolia; Nicaragua; Republic of Moldova; Romania; Sri Lanka; Turkey; Uganda.2 The Committee elected the following officers: Dr R. Wimal Jayantha (Sri Lanka) – Chairman, and Ms Roxana Rotocol (Romania) – Vice-Chairman. The Committee examined the credentials delivered to the Director-General in accordance with Rule 22 of the Rules of Procedure of the World Health Assembly. It noted that the Secretariat had found these credentials to be in conformity with the Rules of Procedure. The credentials of the delegates of the Member States shown at the end of this report were found to be in conformity with the Rules of Procedure as constituting formal credentials; the Committee therefore proposes that the World Health Assembly should recognize their validity. The Committee examined notifications from the Member State listed below, which, while indicating the names of the delegates concerned, could not be considered as constituting formal credentials in accordance with the provisions of the Rules of Procedure. The Committee therefore recommends to the World Health Assembly that the delegates of that Member State be provisionally seated with all rights in the Health Assembly pending the arrival of its formal credentials: Micronesia (Federated States of) Signed by the Officers of the Committee on Credentials, 21 May 2013: Chairman: Dr R.W. -

215Th Commencement 05302017
Greetings from the President May 30, 2017 Dear Graduates: Congratulations! You have reached a most significant milestone in your life. Your hard work, determination, and commitment to your education have been rewarded, and you and your loved ones should take pride in your accomplishments and successes. Hunter College certainly takes pride in you. Your Hunter education has prepared you to meet the challenges of a world that is rapidly changing politically, socially, economically, and technologically. As part of the next generation of thoughtful, responsible, and intelligent leaders, you will make a real difference wherever you apply your knowledge and skills. Endless opportunities await you. As you pursue your goals and move forward with your professional and personal lives, please carry with you Hunter's commitment to community, diversity, and service to otheq . We look forward to hearing great things about you, and we hope you will stay connected to the exciting activities and developments on campus. Please remember Hunter College and know that you will always be part of our family. Best wishes for continued success. f4ivtMSincerely, Jennifer J. Raab President 215th Commencement Exercises Presiding: Jennifer J. Raab President Eija Ayravainen Vice President for Student Affairs and Dean ofStudents Opening Ceremony Qin Lin Bachelor ofArts, '17 Processional President's Party and Members of the Faculty Candidates for Degrees National Anthem Carey Renee Anderson Master ofScience in Education, '17 Bagpiper Nicholas M. Rozak Bachelor ofArts, '07 Greetings Sandra Wilkin Board of Trustees of The City University ofNew York Chika Onyejiukwa Student Member, Board of Trustees of The City University ofNew York Charge to the Graduates President Jennifer J. -

Preliminary Program
Europe’s Past, Present, and Future: Utopias and Dystopias University of Iceland | Reykjavik, Iceland June 22-24, 2020 Preliminary Program This preliminary program is subject to change and CES can not be held liable for any reservation/booking made based on the information provided. Updated on February 7, 2020 Chair: Zsuzsanna Fagyal - University of Illinois at MONDAY, JUNE 22 Urbana-Champaign Participants: [1] Austerity, populism and changing policy Are French people white?: Towards an legitimacy: the European welfare states in understanding of whiteness in Republican France comparative perspective Single Paper 6/22/2020 Jean Beaman - University of California, 9:00 AM to 10:45 AM - Room 14 Santa Barbara Paper Panel Colorblindness and Narratives from the ‘Other’ Chair: Maria Petmesidou - Democritus University of (colorful) France Single Paper Thrace Christina Horvath - University of Bath Ana Guillén - University of Oviedo Participants: ‘Migrants’ or ‘Marseillais?’ The Sociolinguistic Construction of French Minority Youth in Nordic welfare states changes especially in the Marseille light of migration and the financial crisis Single Paper Single Paper Cecile Evers - Pomona College, Bent Greve - Roskilde University Department of Anthropology Jon Kvist - Roskilde University Prototypically non-French: the ‘accent of the The post-Brexit liberal welfare state in Europe: suburbs’ as a cultural construct where next for the United Kingdom? Single Paper Single Paper Zsuzsanna Fagyal - University of Illinois Benjamin Leruth - University of at Urbana-Champaign -

3381-001 Donald Sankey Farner Papers Inventory Accession
UNlVERSllY U BRARIJES w UNIVERSITY of WASHI NGTON Spe ial Colle tions 3784 Donald Sankey Farner papers Inventory Accession No: 3381-001 Special Collections Division University of Washington Libraries Box 352900 Seattle, Washington, 98195-2900 USA (206) 543-1929 This document forms part of the Preliminary Guide to the Donald Sankey Farner Papers. To find out more about the history, context, arrangement, availability and restrictions on this collection, click on the following link: http://digital.lib.washington.edu/findingaids/permalink/FarnerDonaldSUA3381/ Special Collections home page: http://www.lib.washington.edu/specialcollections/ Search Collection Guides: http://digital.lib.washington.edu/findingaids/search DONALDS. FARNER Accession No. 3381-88-21 INVENTORY Box Series Folders Dates 1 GENERAL CORRESPONDENCE A 2 1957-60, 1968, 1970-73,1976-87 Abbott, Ian John 1968 Abelson, Philip H. 1980 Abs, Michael 1981 Academic Press Royalty Statement 1977-79 Adam, Hans 1982 Adkisson, C.S. 1979 Ainley, M.G. 1980, 1985 Akesson, Thomas 1979 Akita, Yasukazu n.d. Aldrich, John W. 1946-58, 1961 1968-72 Alexander Von Humboldt-Stiftung 2 1976-85, 1987 Ali, M.A. 1964 Alvarado, Ronald H. 2 1965, n.d. Alvarez, Bonnie 1970-73 Ameel, Donald J. 1961-65 American Elsevier Publishing Co. n.d. American Express 1977, 1984 Amodon, Dean 1947-51, 1961, 1965-66 Amoroso, E.C. 1961-64, 1974-77 Anderson, Berti! G. n.d. Andrewartha, Herbert G. 1959, 1963 Arcese, Peter 1982 Arnold, Arthur P. 1973-74 Aschoff, Jurgen 1965-83 Ashmole, N. Philip 1969-72 Arvey, M. Dale 1949-73 Assenmacher, Ivan 1960-83 American Society of Zoologists 1983 Audubon Conservation Topics West 1980 Austin, O.L. -

Research and Innovation Annual Report 2018 – 2019
Research and Innovation Annual Report 2018 – 2019 Table of Contents Executive Summary ..................................................................................................................... 3 Outcomes and Impacts of Research ............................................................................................ 4 Outcomes ................................................................................................................................. 4 Impact and Benefits: ................................................................................................................. 6 Research Performance against National Targets ......................................................................... 7 Recruitment .............................................................................................................................. 7 Higher Level Objectives: ....................................................................................................... 7 Performance in Initiating and delivering Clinical Research .................................................... 7 Finance ........................................................................................................................................ 8 Income ..................................................................................................................................... 8 Outcome of funding applications made in 2017/18 ................................................................... 8 Funding applications made in 2018/19 -

Membrane Trafficking in Health and Disease Rebecca Yarwood*, John Hellicar*, Philip G
© 2020. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2020) 13, dmm043448. doi:10.1242/dmm.043448 AT A GLANCE Membrane trafficking in health and disease Rebecca Yarwood*, John Hellicar*, Philip G. Woodman‡ and Martin Lowe‡ ABSTRACT KEY WORDS: Disease, Endocytic pathway, Genetic disorder, Membrane traffic, Secretory pathway, Vesicle Membrane trafficking pathways are essential for the viability and growth of cells, and play a major role in the interaction of cells with Introduction their environment. In this At a Glance article and accompanying Membrane trafficking pathways are essential for cells to maintain poster, we outline the major cellular trafficking pathways and discuss critical functions, to grow, and to accommodate to their chemical how defects in the function of the molecular machinery that mediates and physical environment. Membrane flux through these pathways this transport lead to various diseases in humans. We also briefly is high, and in specialised cells in some tissues can be enormous. discuss possible therapeutic approaches that may be used in the For example, pancreatic acinar cells synthesise and secrete amylase, future treatment of trafficking-based disorders. one of the many enzymes they produce, at a rate of approximately 0.5% of cellular protein mass per hour (Allfrey et al., 1953), while in Schwann cells, the rate of membrane protein export must correlate School of Biological Sciences, Faculty of Biology, Medicine and Health, with the several thousand-fold expansion of the cell surface that University of Manchester, Manchester, M13 9PT, UK. occurs during myelination (Pereira et al., 2012). The population of *These authors contributed equally to this work cell surface proteins is constantly monitored and modified via the ‡Authors for correspondence ([email protected]; endocytic pathway. -

Download PDF (90.8
Index Aboltin, V.Y. 95, 98, 102 Annan, N. 67, 206 Ackroyd, E. 40 applied economics acts (U.K.) definition 4, 5–6, 12, 21, 78, 127–8 Distribution of Industry (Industrial handling data 7–8, 189 Finance) Act (1958) 169 history 1, 5–6 Distribution of Industry Act (1945) mathematics 40, 135, 204 168 public policy 1, 12, 40, 65, 86, 151, Education Act (1944) 157 191, 202 Employment Act (1946) 161 realism 13–14 Employment and Training Act Armstrong, H.W. 169, 206 (1973) 184 Ashby, E. 162 Finance Act (1963) 169 Association of University Teachers 79 Government of India Act (1935) 105 Atkinson, R. 160 Industrial Development Act (1966) Attlee, C. 85 169, 183 Ayres, C. 7 Industry Act (1975) 184 Labour Exchanges Act (1909) 167 Backhouse, R.E. 5–6, 206 Local Employment Act (1960) 169, Baker, P.N. 103 183 Balogh, T. 36, 156, 162 Local Employment Act (1963) 183 Banda, H. 109–10 Oxford University Act (1854) 61 Bank of England 27, 152, 154, 161 Representation of People Act (1948) Barber, A.P.L. 191 65 Barr, N. 123, 125, 206 Special Areas (Development and Barro, R.J. 188, 206 Improvement) Act (1934) 168 Baumol, W.J. 206 Special Areas Amendment Act Beeching, R. 76 (1937) 168 Beesley, M. 76 Adler, J.H. 57, 206 Beheiry, M. 96 Advisory Commission on the Review Bellamy, R. 69 of the Constitution of Rhodesia Beloff, M. 8, 155, 206 and Nyasaland 106 Benham, F.C.37, 60, 206 Aiken, J. 158 Benoit, E. 98, 103, 114 Akerlof, G. -

Download Complete Issue
ISLAND STUDIES JOURNAL Volume 13 | Issue 2 | November 2018 islandstudies.ca ISSN: 1715-2593 ISLAND STUDIES JOURNAL Island Studies Journal is a peer-reviewed open access journal published by the University of Prince Edward Island’s Institute of Island Studies. https://www.islandstudies.ca Executive editor Adam Grydehøj (University of Prince Edward Island, Canada) Editorial board Laurie Brinklow (University of Prince Edward Island, Canada) Lead Copyeditor Gordon Cooke (Memorial University of Newfoundland, Canada) Deputy Editor for Business & Economics Scholarship Lisa Fletcher (University of Tasmania, Australia) Deputy Editor for Book Reviews Ilan Kelman (University College London, UK) Deputy Editor for Excellence & Disciplines Ulunnguaq Markussen (Ilisimatusarfik/University of Greenland, Kalaallit Nunaat) Deputy Editor for Indigenous Research & Outreach G. Edward MacDonald (University of Prince Edward Island, Canada) Deputy Editor for Institutional Relations Maeve McCusker (Queen's University Belfast, Northern Ireland) Deputy Editor for Literature Scholarship Yaso Nadarajah (RMIT University, Australia) Deputy Editor for Diversity & Regions Jan Petzold (Alfred Wegner Institute for Polar and Marine Research, Germany) Deputy Editor for Climate Change and Environment Scholarship Evangelia Papoutsaki (Unitec Institute of Technology, New Zealand) Deputy Editor for Digital Communications Elaine Stratford (University of Tasmania, Australia) Deputy Editor for Policy Impact Godfrey Baldacchino (University of Malta, Malta) Editor Emeritus Editorial -
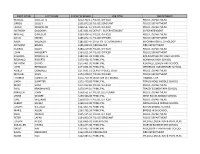
First Name Last Name Salary & Wages Job Title Department
FIRST NAME LAST NAME SALARY & WAGES JOB TITLE DEPARTMENT RUSSELL OUELLETTE $212,702.11 POLICE OFFICER POLICE DEPARTMENT GREGG SCULLY $205,873.26 POLICE SERGEANT POLICE DEPARTMENT JAVIER MOGOLLON $198,547.61 POLICE OFFICER POLICE DEPARTMENT ANTHONY DADDONA $197,691.02 DEPUTY SUPERINTENDENT SUPERINTENDENT MICHAEL DIMEGLIO $190,024.41 POLICE OFFICER POLICE DEPARTMENT DAVID NIEVES $185,842.41 POLICE OFFICER POLICE DEPARTMENT RALPH VALENZISI $185,823.47 CHIEF OF TECH/INN/PART INFORMATION TECHNOLOGY ANTHONY BRUNO $185,038.39 FIREFIGHTER FIRE DEPARTMENT GEORGE DALEY $184,219.06 POLICE OFFICER POLICE DEPARTMENT JOHN HAGGERTY $184,011.94 POLICE OFFICER POLICE DEPARTMENT SUZANNE KOROSHETZ $180,451.94 PRINCIPAL BRIEN MCMAHON HIGH SCHOOL REGINALD ROBERTS $179,428.49 PRINCIPAL NORWALK HIGH SCHOOL ANTHONY DITRIO $175,401.92 PRINCIPAL KENDALL ELEMENTARY SCHOOL JOHN REYNOLDS $174,651.99 PRINCIPAL JEFFERSON ELEMENTARY SCHOOL ASHLEY GONZALEZ $173,691.52 DEPUTY POLICE CHIEF POLICE DEPARTMENT MICHAEL SILVA $173,378.57 POLICE OFFICER POLICE DEPARTMENT THOMAS HAMILTON $172,760.39 DIRECTOR OF FINANCE FINANCE CITY LINDA SUMPTER $172,449.88 PRINCIPAL PONUS RIDGE MIDDLE SCHOOL MARK SUDA $171,022.82 POLICE OFFICER POLICE DEPARTMENT PAUL KRASNAVAGE $170,344.01 PRINCIPAL TRACEY ELEMENTARY SCHOOL PRAVEEN JOHN $169,551.67 POLICE LIEUTENANT POLICE DEPARTMENT LYNNE MOORE $169,366.08 PRINCIPAL WEST ROCKS MIDDLE SCHOOL CARL WILLIAMS $169,079.77 POLICE OFFICER POLICE DEPARTMENT ALBERT SACKEY $168,212.00 PRINCIPAL NATHAN HALE MIDDLE SCHOOL JOSEPH VELLUCCI $167,865.92 PRINCIPAL -
ECCOMAS Congress 2016 PROGRAMME
European Community on Computational Methods in Applied Sciences ECCOMAS Congress 2016 VII European Congress on Computational Methods in Applied Sciences and Engineering June 5-10, Crete, Greece PROGRAMME Conference Secretariat: Institute of Structural Analysis and Antiseismic Research National Technical University of Athens, Greece ECCOMAS Congress 2016 7th European Congress on Computational Methods in Applied Sciences and Engineering 5-10 June 2016, Crete Island, Greece Programme Institute of Structural Analysis and Antiseismic Research School of Civil Engineering National Technical University of Athens Table of Contents Greetings ……....…………………………………………….………………………………………….…… 3 Scientific Programme Overview ………………………………………………………….………… 6 Floor Plans ……………….…………………………………………………………………………….……… 16 ECCOMAS Congress 2016 Organizing Committees ……………….…….…………….…… 19 ECCOMAS Congress 2016 Scientific Committees ………………….…….…………….…… 20 Congress Venue ……………………………………………………………………………………………. 22 Transportation ….…………..………………………………………………………………………..….... 23 Congress Information ………..……………………………………………….……….……………….. 24 Social Events …………………...……………………………………………………….……………..….. 26 Associations Meetings …….……………………………………………………..…………………….. 28 Plenary Lecturers …………..………………………..………………………………………………..….. 29 Semi-Plenary Lecturers ......………………………………………………………………………..….. 30 Keynote Lecturers ….………..…………………………………………………………………….…….. 34 Minisymposia ….…………..……………………….…………………………………..………….…….... 42 Young Investigators Minisymposium ….…………..…………………..…………………..…… 52 ECCOMAS Olympiad -
Molecular Interactions Between Childhood Acute Lymphoblastic Leukaemia Cells and the Bone Marrow Microenvironment
Molecular Interactions Between Childhood Acute Lymphoblastic Leukaemia Cells And The Bone Marrow Microenvironment Ana Markovic A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Children’s Cancer Institute Australia for Medical Research School of Women’s and Children’s Health THE UNIVERSITY OF NEW SOUTH WALES 2009 THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: MARKOVIC First name: ANA Other name/s: Abbreviation for degree as given in the University calendar: PhD School: Women’s & Children’s Health Faculty: MEDICINE Title: Molecular Interactions Between Childhood Acute Lymphoblastic Leukaemia Cells And The Bone Marrow Microenvironment. Abstract 350 words maximum Acute lymphoblastic leukaemia (ALL) is the most common cause of death from disease in children. Whilst cure rates over the last 30 years have drastically improved, children that do go on and relapse have a very poor prognosis. Additionally, the ones that do survive can have significant long term side effects from existing treatments. Understanding the molecular mechanisms of the relationship between leukaemia and its microenvironment is essential for the identification of novel targets for treatment and/or the manipulation of existing treatments. Our previous work has established a panel of childhood ALL xenografts from patient biopsies in NOD/SCID mice. Several samples secrete vascular endothelial growth factor (VEGF), an integral component of neovascularisation and normal haematopoiesis, and express FMS-like tyrosine kinase-3 (FLT-3), a receptor tyrosine kinase, which plays an essential role in regulating normal haematopoiesis. This thesis builds on previous work by examining the relationship between VEGF and FLT-3, two widely, yet independently studied molecules in leukaemia, with the aberrant expression of either having adverse outcomes for patients. -

Faculty of Commerce (Ceremony 2)
FACULTY OF COMMERCE (CEREMONY 2) ORDER OF PROCEEDINGS Academic Procession. (The congregation is requested to stand as the procession enters the hall) The Presiding Officer will constitute the congregation. The National Anthem. The University Statement of Dedication will be read by a representative of the SRC. Musical Item. Welcome by the Master of Ceremonies. The Master of Ceremonies will introduce the guest speaker. Address by the guest speaker. The graduands will be presented to the Presiding Officer by the Dean of Commerce. The Presiding Officer will congratulate the new graduates. The Master of Ceremonies will make closing announcements and invite the congregation to stand. The Presiding Officer will dissolve the congregation. The procession, including the new graduates, will leave the hall. (The congregation is requested to remain standing until the procession has left the hall.) 1 NATIONAL ANTHEM Nkosi sikelel’ iAfrika Maluphakanyisw’ uphondolwayo, Yizwa imithandazo yethu, Nkosi sikelela, thina lusapho lwayo. Morena boloka etjhaba sa heso, O fedise dintwa la matshwenyeho, O se boloke, O se boloke setjhaba sa heso, Setjhaba sa South Afrika – South Afrika. Uit die blou van onse hemel, Uit die diepte van ons see, Oor ons ewige gebergtes, Waar die kranse antwoord gee, Sounds the call to come together, And united we shall stand, Let us live and strive for freedom, In South Africa our land. 2 DISTINCTIONS IN THE FACULTY OF COMMERCE The BCom and BBusSc degrees may be awarded with distinction in a subject, where the student achieves a weighted average of at least 75% in a group of courses in, or related to, that subject (in some cases the average mark required is 80%) in the degree, where the student achieves a weighted average of 80% in all courses.