1 Large-Scale Maps of Variable Infection Efficiencies in Aquatic Bacteroidetes Phage
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extreme Cold Environments: a Suitable Niche for Selection of Novel Psychrotrophic Microbes for Biotechnological Applications
Editorial Adv Biotech & Micro Volume 2 Issue 2 - February 2017 Copyright © All rights are reserved by Ajar Nath Yadav DOI: 10.19080/AIBM.2017.02.555584 Extreme Cold Environments: A Suitable Niche for Selection of Novel Psychrotrophic Microbes for Biotechnological Applications Ajar Nath Yadav1*, Priyanka Verma2, Vinod Kumar1, Shashwati Ghosh Sachan3 and Anil Kumar Saxena4 1Department of Biotechnology, Eternal University, India 2Department of Microbiology, Eternal University, India 3Department of Bio-Engineering, Birla Institute of Technology, India 4ICAR-National Bureau of Agriculturally Important Microorganisms, India Submission: January 27, 2017; Published: February 06, 2017 *Corresponding author: Ajar Nath Yadav, Assistant professor, Department of Biotechnology, Akal College of Agriculture, Eternal University, India, Tel: ; Email: Editorial several reports on whole genome sequences of novel and The microbiomes of cold environments are of particular potential psychrotrophic microbes [26,27]. importance in global ecology since the majority of terrestrial and aquatic ecosystems of our planet are permanently or The novel species of psychrotrophic microbes have been seasonally submitted to cold temperatures. Earth is primarily a isolated worldwide and reported from different domain cold, marine planet with 90% of the ocean’s waters being at 5°C archaea, bacteria and fungi which included members of phylum or lower. Permafrost soils, glaciers, polar sea ice, and snow cover Actinobacteria, Proteobacteria, Bacteroidetes, Basidiomycota, make up 20% of the Earth’s surface environments. Microbial Firmicutes and Euryarchaeota [7-25]. Along with novel species communities under cold habitats have been undergone the of psychrotrophic microbes, some microbial species including physiological adaptations to low temperature and chemical Arthrobacter nicotianae, Brevundimonas terrae, Paenibacillus stress. -

Discovering Novel Enzymes by Functional Screening of Plurigenomic
Microbiological Research 186–187 (2016) 52–61 Contents lists available at ScienceDirect Microbiological Research j ournal homepage: www.elsevier.com/locate/micres Discovering novel enzymes by functional screening of plurigenomic libraries from alga-associated Flavobacteriia and Gammaproteobacteria a,∗ b a a Marjolaine Martin , Marie Vandermies , Coline Joyeux , Renée Martin , c c a Tristan Barbeyron , Gurvan Michel , Micheline Vandenbol a Microbiology and Genomics Unit, Gembloux Agro-Bio Tech, University of Liège, Passage des Déportés 2, 5030 Gembloux, Belgium b Microbial Processes and Interactions, Gembloux Agro-Bio Tech, University of Liège, Passage des Déportés 2, 5030 Gembloux, Belgium c Sorbonne Université, UPMC Univ Paris 06, CNRS, UMR 8227, Integrative Biology of Marine Models, Station Biologique de Roscoff, CS 90074, F-29688 Roscoff cedex, Bretagne, France a r t i c l e i n f o a b s t r a c t Article history: Alga-associated microorganisms, in the context of their numerous interactions with the host and the Received 4 February 2016 complexity of the marine environment, are known to produce diverse hydrolytic enzymes with original Received in revised form 17 March 2016 biochemistry. We recently isolated several macroalgal-polysaccharide-degrading bacteria from the sur- Accepted 22 March 2016 face of the brown alga Ascophyllum nodosum. These active isolates belong to two classes: the Flavobacteriia Available online 24 March 2016 and the Gammaproteobacteria. In the present study, we constructed two “plurigenomic” (with multi- ple bacterial genomes) libraries with the 5 most interesting isolates (regarding their phylogeny and Keywords: their enzymatic activities) of each class (Fv and Gm libraries). Both libraries were screened for diverse Alga-associated microflora hydrolytic activities. -

The Cell Organization Underlying Structural Colour Is Involved in Flavobacterium IR1 Predation
The ISME Journal (2020) 14:2890–2900 https://doi.org/10.1038/s41396-020-00760-6 ARTICLE The cell organization underlying structural colour is involved in Flavobacterium IR1 predation 1 2 1,3 1 Raditijo Hamidjaja ● Jérémie Capoulade ● Laura Catón ● Colin J. Ingham Received: 29 March 2020 / Revised: 14 August 2020 / Accepted: 24 August 2020 / Published online: 1 September 2020 © The Author(s) 2020. This article is published with open access Abstract Flavobacterium IR1 is a gliding bacterium with a high degree of colonial organization as a 2D photonic crystal, resulting in vivid structural coloration when illuminated. Enterobacter cloacae B12, an unrelated bacterium, was isolated from the brown macroalga Fucus vesiculosus from the same location as IR1. IR1 was found to be a predator of B12. A process of surrounding, infiltration, undercutting and killing of B12 supported improved growth of IR1. A combination of motility and capillarity facilitated the engulfment of B12 colonies by IR1. Predation was independent of illumination. Mutants of IR1 that formed photonic crystals less effectively than the wild type were reduced in predation. Conversely, formation of a photonic crystal was not advantageous in resisting predation by Rhodococcus spp. PIR4. These observations suggest that the 1234567890();,: 1234567890();,: organization required to create structural colour has a biological function (facilitating predation) but one that is not directly related to the photonic properties of the colony. This work is the first experimental evidence supporting a role for this widespread type of cell organization in the Flavobacteriia. Introduction to highly repeated arrays of cells which can assemble rapidly to form a 2D photonic crystal (2DPC) [1], a form of Flavobacterium IR1 is a gliding bacterium that can spread structural colour (SC), which is distinct in mechanism from over hydrated surfaces [1]. -

Diversity of Antibiotic-Active Bacteria Associated with the Brown Alga Laminaria Saccharina from the Baltic Sea
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by OceanRep Mar Biotechnol (2009) 11:287–300 DOI 10.1007/s10126-008-9143-4 ORIGINAL ARTICLE Diversity of Antibiotic-Active Bacteria Associated with the Brown Alga Laminaria saccharina from the Baltic Sea Jutta Wiese & Vera Thiel & Kerstin Nagel & Tim Staufenberger & Johannes F. Imhoff Received: 28 January 2008 /Accepted: 1 September 2008 /Published online: 15 October 2008 # Springer Science + Business Media, LLC 2008 Abstract Bacteria associated with the marine macroalga (Bartsch et al. 2008). Epiphytic bacteria have been studied Laminaria saccharina, collected from the Kiel Fjord (Baltic by microscopic methods (Corre and Prieur 1990) and by Sea, Germany), were isolated and tested for antimicrobial genetic and cultivation approaches. Bacterial cell numbers activity. From a total of 210 isolates, 103 strains inhibited of up to 107 colony-forming units (CFU) per centimeter the growth of at least one microorganism from the test squared were reported for Laminaria digitata from the panel including Gram-negative and Gram-positive bacteria coast of Brittany (France) and Laminaria pallida on the as well as a yeast. Most common profiles were the Bengal upwelling region of southern Africa (Corre and inhibition of Bacillus subtilis only (30%), B. subtilis and Prieur 1990; Mazure and Field 1980). However, the Staphylococcus lentus (25%), and B. subtilis, S. lentus, and interactions between members of the epiphytic and endo- Candida albicans (11%). In summary, the antibiotic-active phytic communities and the relationships between these isolates covered 15 different activity patterns suggesting communities and Laminaria spp. -

Analysis of Metagenome-Assembled Viral Genomes from the Human Gut Reveals Diverse Putative Crass-Like Phages with Unique Genomic Features
ARTICLE https://doi.org/10.1038/s41467-021-21350-w OPEN Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features Natalya Yutin1, Sean Benler1, Sergei A. Shmakov1, Yuri I. Wolf 1, Igor Tolstoy1, Mike Rayko 2, ✉ Dmitry Antipov2, Pavel A. Pevzner 3 & Eugene V. Koonin 1 1234567890():,; CrAssphage is the most abundant human-associated virus and the founding member of a large group of bacteriophages, discovered in animal-associated and environmental meta- genomes, that infect bacteria of the phylum Bacteroidetes. We analyze 4907 Circular Metagenome Assembled Genomes (cMAGs) of putative viruses from human gut micro- biomes and identify nearly 600 genomes of crAss-like phages that account for nearly 87% of the DNA reads mapped to these cMAGs. Phylogenetic analysis of conserved genes demonstrates the monophyly of crAss-like phages, a putative virus order, and of 5 branches, potential families within that order, two of which have not been identified previously. The phage genomes in one of these families are almost twofold larger than the crAssphage genome (145-192 kilobases), with high density of self-splicing introns and inteins. Many crAss-like phages encode suppressor tRNAs that enable read-through of UGA or UAG stop- codons, mostly, in late phage genes. A distinct feature of the crAss-like phages is the recurrent switch of the phage DNA polymerase type between A and B families. Thus, comparative genomic analysis of the expanded assemblage of crAss-like phages reveals aspects of genome architecture and expression as well as phage biology that were not apparent from the previous work on phage genomics. -
Comparative Genomic Analysis of Flavobacteriaceae: Insights Into Carbohydrate Metabolism, Gliding Motility and Secondary Metabolite Biosynthesis
Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. Asimenia Gavriilidou ( [email protected] ) Wageningen Universiteit en Research https://orcid.org/0000-0003-0204-2223 Johanna Gutleben Wageningen Universiteit en Research Dennis Versluis Wageningen Universiteit en Research https://orcid.org/0000-0001-8430-4500 Francesca Forgiarini Wageningen Universiteit en Research Mark WJ van Passel Wageningen Universiteit en Research https://orcid.org/0000-0001-6519-641X Colin J Ingham Hoekmine BV https://orcid.org/0000-0003-1508-019X Hauke Smidt Wageningen Universiteit en Research https://orcid.org/0000-0002-6138-5026 Detmer Sipkema Wageningen Universiteit en Research https://orcid.org/0000-0003-3836-219X Research article Keywords: Comparative genomics, Flavobacteriaceae, marine, host-associated, free-living Posted Date: March 3rd, 2020 DOI: https://doi.org/10.21203/rs.3.rs-15750/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on August 20th, 2020. See the published version at https://doi.org/10.1186/s12864-020-06971-7. Page 1/33 Abstract Background Members of the bacterial family Flavobacteriaceae are widely distributed in the marine environment and often found associated with algae, sh, detritus or marine invertebrates. Yet, little is known about the characteristics that drive their ubiquity in diverse ecological niches. Here, we provide an overview of functional traits common to taxonomically diverse members of the family Flavobacteriaceae from different environmental sources, with a focus on the Marine clade. We include seven newly sequenced marine sponge-derived strains that were also tested for gliding motility and antimicrobial activity. -

Glitter-Like Iridescence Within the Bacteroidetes Especially Cellulophaga Spp.: Optical Properties and Correlation with Gliding Motility
Glitter-Like Iridescence within the Bacteroidetes Especially Cellulophaga spp.: Optical Properties and Correlation with Gliding Motility Betty Kientz1, Adrien Ducret2, Stephen Luke3, Peter Vukusic3,Taˆm Mignot2, Eric Rosenfeld1* 1 UMR 7266 CNRS Littoral Environnement et Socie´te´s, University of La Rochelle, La Rochelle, France, 2 UMR 7283 CNRS Laboratoire de Chimie Bacte´rienne, Institut de Microbiologie de la Me´diterrane´e, University of Aix-Marseille, Marseille, France, 3 School of Physics, University of Exeter, Exeter, United Kingdom Abstract Iridescence results from structures that generate color. Iridescence of bacterial colonies has recently been described and illustrated. The glitter-like iridescence class, created especially for a few strains of Cellulophaga lytica, exhibits an intense iridescence under direct illumination. Such color appearance effects were previously associated with other bacteria from the Bacteroidetes phylum, but without clear elucidation and illustration. To this end, we compared various bacterial strains to which the iridescent trait was attributed. All Cellulophaga species and additional Bacteroidetes strains from marine and terrestrial environments were investigated. A selection of bacteria, mostly marine in origin, were found to be iridescent. Although a common pattern of reflected wavelengths was recorded for the species investigated, optical spectroscopy and physical measurements revealed a range of different glitter-like iridescence intensity and color profiles. Importantly, gliding motility was found to be a common feature of all iridescent colonies. Dynamic analyses of ‘‘glitter’’ formation at the edges of C. lytica colonies showed that iridescence was correlated with layer superposition. Both gliding motility, and unknown cell- to-cell communication processes, may be required for the establishment, in time and space, of the necessary periodic structures responsible for the iridescent appearance of Bacteroidetes. -

Twelve Previously Unknown Phage Genera Are Ubiquitous in Global Oceans
Twelve previously unknown phage genera are ubiquitous in global oceans Karin Holmfeldta,1,2, Natalie Solonenkoa, Manesh Shahb, Kristen Corrierb, Lasse Riemannc, Nathan C. VerBerkmoesb,3, and Matthew B. Sullivana,1 aDepartment of Ecology and Evolutionary Biology, University of Arizona, Tucson, AZ 85721; bChemical Science Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831; and cDepartment of Biology, University of Copenhagen, 3000 Helsingor, Denmark Edited by James L. Van Etten, University of Nebraska–Lincoln, Lincoln, NE, and approved June 17, 2013 (received for review April 2, 2013) Viruses are fundamental to ecosystems ranging from oceans to (7) and deep (8) oceans and even the human gut. In humans, humans, yet our ability to study them is bottlenecked by the lack Bacteroidetes comprise 30% of the gut microbiota and play im- of ecologically relevant isolates, resulting in “unknowns” dominat- portant roles for fat storage (9) and the immune system (10). In ing culture-independent surveys. Here we present genomes from the oceans, Bacteroidetes is the third most abundant bacterial 31 phages infecting multiple strains of the aquatic bacterium Cel- phylum (7, 8), and there these bacteria are active in degrading lulophaga baltica (Bacteroidetes) to provide data for an under- biopolymers (11) and involved in recycling of phytoplankton represented and environmentally abundant bacterial lineage. bloom-related organic matter (12). Comparative genomics delineated 12 phage groups that (i) each Here we present 31 genomes and 13 representative structural proteomes of Bacteroidetes phages isolated by using 17 Cellulo- represent a new genus, and (ii) represent one novel and four well- phaga host strains. This genomic and proteomic information known viral families. -
The Cultivable Surface Microbiota of the Brown Alga Ascophyllum Nodosum Is Enriched in Macroalgal- Polysaccharide-Degrading Bacteria
ORIGINAL RESEARCH published: 24 December 2015 doi: 10.3389/fmicb.2015.01487 The Cultivable Surface Microbiota of the Brown Alga Ascophyllum nodosum is Enriched in Macroalgal- Polysaccharide-Degrading Bacteria Marjolaine Martin 1*, Tristan Barbeyron 2, Renee Martin 1, Daniel Portetelle 1, Gurvan Michel 2 and Micheline Vandenbol 1 1 Microbiology and Genomics Unit, Gembloux Agro-Bio Tech, University of Liège, Gembloux, Belgium, 2 Sorbonne Université, UPMC, Centre National de la Recherche Scientifique, UMR 8227, Integrative Biology of Marine Models, Roscoff, France Bacteria degrading algal polysaccharides are key players in the global carbon cycle and in algal biomass recycling. Yet the water column, which has been studied largely by metagenomic approaches, is poor in such bacteria and their algal-polysaccharide-degrading enzymes. Even more surprisingly, the few published studies on seaweed-associated microbiomes have revealed low abundances of such bacteria and their specific enzymes. However, as macroalgal cell-wall polysaccharides Edited by: do not accumulate in nature, these bacteria and their unique polysaccharidases must Olga Lage, not be that uncommon. We, therefore, looked at the polysaccharide-degrading activity University of Porto, Portugal of the cultivable bacterial subpopulation associated with Ascophyllum nodosum. From Reviewed by: Torsten Thomas, A. nodosum triplicates, 324 bacteria were isolated and taxonomically identified. Out of The University of New South Wales, these isolates, 78 (∼25%) were found to act on at least one tested algal polysaccharide Australia (agar, ι- or κ-carrageenan, or alginate). The isolates “active” on algal-polysaccharides Suhelen Egan, The University of New South Wales, belong to 11 genera: Cellulophaga, Maribacter, Algibacter, and Zobellia in the class Australia Flavobacteriia (41) and Pseudoalteromonas, Vibrio, Cobetia, Shewanella, Colwellia, *Correspondence: Marinomonas, and Paraglaceciola in the class Gammaproteobacteria (37). -

Cellulophaga Pacifica Sp. Nov
International Journal of Systematic and Evolutionary Microbiology (2004), 54, 609–613 DOI 10.1099/ijs.0.02737-0 Cellulophaga pacifica sp. nov. Olga I. Nedashkovskaya,1 Makoto Suzuki,2 Anatoly M. Lysenko,3 Cindy Snauwaert,4 Marc Vancanneyt,4 Jean Swings,4 Mikhail V. Vysotskii5 and Valery V. Mikhailov1 Correspondence 1Pacific Institute of Bioorganic Chemistry of the Far-Eastern Branch of the Russian Academy Olga I. Nedashkovskaya of Sciences, Pr. 100 Let Vladivostoku 159, 690022, Vladivostok, Russia [email protected] 2Tokyo Research Laboratories, Kyowa Hakko Kogyo Company Ltd, 3-6-6 Asahi-machi, or Machida-shi, Tokyo 194-8533, Japan [email protected] 3Institute of Microbiology of the Russian Academy of Sciences, Pr. 60 let October 7/2, Moscow, 117811, Russia 4BCCM/LMG Bacteria Collection, Laboratory of Microbiology, Faculty of Sciences, Ghent University, Ledeganckstraat 35, B 9000 Ghent, Belgium 5Institute of Marine Biology of the Far-Eastern Branch of the Russian Academy of Sciences, Pal’chevskogo St 17, 690032, Vladivostok, Russia Three marine, heterotrophic, aerobic, agarolytic, pigmented and gliding bacteria were isolated in June 2000 from a sea water sample that was collected in the Gulf of Peter the Great, Sea of Japan, and analysed in a polyphasic taxonomic study. 16S rDNA sequence analysis indicated that strains KMM 3664T, KMM 3669 and KMM 3915 were members of the family Flavobacteriaceae. Based on phenotypic, chemotaxonomic, genotypic and phylogenetic data, the isolates were classified in the genus Cellulophaga as members of a novel species, Cellulophaga pacifica sp. nov. The type strain is KMM 3664T (=JCM 11735T=LMG 21938T). The genus Cellulophaga was proposed by Johansen et al. -

Discovering Novel Enzymes By
DISCOVERING NOVEL ENZYMES BY FUNCTIONAL SCREENING OF PLURIGENOMIC LIBRARIES FROM ALGA-ASSOCIATED FLAVOBACTERIIA AND GAMMAPROTEOBACTERIA Marjolaine Martin, Marie Vandermies, Coline Joyeux, Renee Martin, Tristan Barbeyron, Gurvan Michel, Micheline Vandenbol To cite this version: Marjolaine Martin, Marie Vandermies, Coline Joyeux, Renee Martin, Tristan Barbeyron, et al.. DISCOVERING NOVEL ENZYMES BY FUNCTIONAL SCREENING OF PLURIGENOMIC LI- BRARIES FROM ALGA-ASSOCIATED FLAVOBACTERIIA AND GAMMAPROTEOBACTERIA. Microbiological Research, Elsevier, 2016, 186-187, pp.52-61. 10.1016/j.micres.2016.03.005. hal- 02137941 HAL Id: hal-02137941 https://hal.archives-ouvertes.fr/hal-02137941 Submitted on 23 May 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. *Manuscript 1 DISCOVERING NOVEL ENZYMES BY FUNCTIONAL SCREENING OF PLURIGENOMIC 2 LIBRARIES FROM ALGA-ASSOCIATED FLAVOBACTERIIA AND GAMMAPROTEOBACTERIA 3 4 Marjolaine Martin1*, Marie Vandermies2, Coline Joyeux1, Renée Martin1, Tristan Barbeyron3, 5 Gurvan Michel3, Micheline Vandenbol1 6 1 Microbiology and -
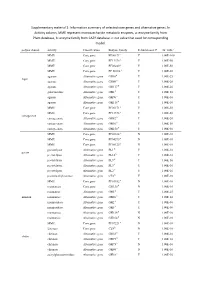
Information Summary of Selected Core Genes and Alternative Genes. in Activity Column, MME Represent Mo
Supplementary material 1: Information summary of selected core genes and alternative genes. In Activity column, MME represent monosaccharide metabolic enzymes, a: enzyme family from Pfam database, b: enzyme family from CAZY database. c: cut value that used for corresponding model. polysaccharide activity Classification Enzyme family Rebuild model? cut_valuec MME Core gene PF00171a Y 1.00E-100 MME Core gene PF13378 a Y 1.00E-50 MME Core gene PF08240 a Y 1.00E-50 MME Core gene PF 00106 a Y 1.00E-50 agarase Alternative gene GH16b Y 1.00E-25 Agar agarase Alternative gene GH86 b Y 1.00E-20 agarase Alternative gene GH117 b Y 1.00E-20 galactosidase Alternative gene GH2 b Y 1.00E-50 agarase Alternative gene GH96 b Y 1.00E-10 agarase Alternative gene GH118 b Y 1.00E-10 MME Core gene PF00171 a Y 1.00E-50 MME Core gene PF13378 a Y 1.00E-50 carrageenan carrageenase Alternative gene GH82 b Y 1.00E-20 carrageenase Alternative gene GH16 b Y 1.00E-50 carrageenase Alternative gene GH150 b Y 1.00E-50 MME Core gene PF02614 a N 1.00E-10 MME Core gene PF04295 a N 1.00E-10 MME Core gene PF08125 a N 1.00E-10 pectatelyase Alternative gene PL1 b Y 1.00E-10 pectin pectatelyase Alternative gene PL10 b Y 1.00E-10 pectatelyase Alternative gene PL9 b Y 1.00E-50 pectatelyase Alternative gene PL3 b Y 1.00E-10 pectatelyase Alternative gene PL2 b Y 1.00E-10 pectinmethylesterase Alternative gene CE8 b Y 1.00E-10 MME Core gene PF01182 a N 1.00E-10 mannanase Core gene GH130 b N 1.00E-10 mannanase Alternative gene GH5 b Y 1.00E-25 mannan mannanase Alternative gene GH26 b