Chronic Inhibition of STAT3/STAT5 in Treatment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drosophila STING Protein Has a Role in Lipid Metabolism
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.04.429825; this version posted February 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Drosophila STING protein has a role in lipid metabolism. Katarina Akhmetova, Maxim Balasov and Igor Chesnokov*. Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, School of Medicine, 720 20th Street South, Birmingham, Alabama 35294 Running head: The role of dSTING in metabolism. *Corresponding Author. Mailing address: Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, School of Medicine, 720 20th Street South, Birmingham, Alabama 35294. Phone: (205) 934- 6974. Fax: (205) 934-0758. E-mail: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2021.02.04.429825; this version posted February 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ABSTRACT Stimulator of interferon genes (STING) plays an important role in innate immunity by controlling type I interferon response against invaded pathogens. In this work we describe a direct but previously unknown role of STING in lipid metabolism in Drosophila. Flies with STING deletion are sensitive to starvation and oxidative stress, have reduced lipid storage and downregulated expression of lipid metabolism genes. -

Transdifferentiation of Human Mesenchymal Stem Cells
Transdifferentiation of Human Mesenchymal Stem Cells Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Julius-Maximilians-Universität Würzburg vorgelegt von Tatjana Schilling aus San Miguel de Tucuman, Argentinien Würzburg, 2007 Eingereicht am: Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Martin J. Müller Gutachter: PD Dr. Norbert Schütze Gutachter: Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am: Hiermit erkläre ich ehrenwörtlich, dass ich die vorliegende Dissertation selbstständig angefertigt und keine anderen als die von mir angegebenen Hilfsmittel und Quellen verwendet habe. Des Weiteren erkläre ich, dass diese Arbeit weder in gleicher noch in ähnlicher Form in einem Prüfungsverfahren vorgelegen hat und ich noch keinen Promotionsversuch unternommen habe. Gerbrunn, 4. Mai 2007 Tatjana Schilling Table of contents i Table of contents 1 Summary ........................................................................................................................ 1 1.1 Summary.................................................................................................................... 1 1.2 Zusammenfassung..................................................................................................... 2 2 Introduction.................................................................................................................... 4 2.1 Osteoporosis and the fatty degeneration of the bone marrow..................................... 4 2.2 Adipose and bone -

Proteoglycan Form and Function: a Comprehensive Nomenclature of Proteoglycans
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Matrix Biol Manuscript Author . Author manuscript; Manuscript Author available in PMC 2016 May 06. Published in final edited form as: Matrix Biol. 2015 March ; 42: 11–55. doi:10.1016/j.matbio.2015.02.003. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans Renato V. Iozzo1 and Liliana Schaefer2 1Department of Pathology, Anatomy and Cell Biology and the Cancer Cell Biology and Signaling Program, Kimmel Cancer Center, Sidney Kimmel Medical College at Thomas Jefferson University, Philadelphia, PA 19107, USA 2Pharmazentrum Frankfurt/ZAFES, Institut für Allgemeine Pharmakologie und Toxikologie, Klinikum der Goethe-Universität Frankfurt am Main, Frankfurt am Main, Germany Abstract We provide a comprehensive classification of the proteoglycan gene families and respective protein cores. This updated nomenclature is based on three criteria: Cellular and subcellular location, overall gene/protein homology, and the utilization of specific protein modules within their respective protein cores. These three signatures were utilized to design four major classes of proteoglycans with distinct forms and functions: the intracellular, cell-surface, pericellular and extracellular proteoglycans. The proposed nomenclature encompasses forty-three distinct proteoglycan-encoding genes and many alternatively-spliced variants. The biological functions of these four proteoglycan families are critically assessed in development, cancer and angiogenesis, and in various acquired and genetic diseases where their expression is aberrant. Keywords Proteoglycan; Glycosaminoglycan; Cancer growth; Angiogenesis; Growth factor modulation Introduction It has been nearly 20 years since the original publication of a comprehensive classification of proteoglycan gene families [1]. For the most part, these classes have been widely accepted. However, a broad and current taxonomy of the various proteoglycan gene families and their products is not available. -

University of Cincinnati
UNIVERSITY OF CINCINNATI January 3, 2002 I, Arthur M. Barrie, III, hereby submit this as part of the requirements for the degree of: Doctorate of Philosophy (Ph.D.) in: the Graduate Program in Molecular and Developmental Biology It is entitled: " The Role and Regulation of the Anti-Inflammatory Mouse Apolipoprotein J Gene" Approved by: Dr. Bruce Aronow, Ph.D. Dr. John Bissler, M.D. Dr. Robert Colbert, M.D., Ph.D. Dr. Sandra Degen, Ph.D Dr. Jun Ma, Ph.D. THE ROLE AND REGULATION OF THE ANTI-INFLAMMATORY MOUSE APOLIPOPROTEIN J GENE A dissertation submitted to the Division of Research and Advanced Studies of the University of Cincinnati in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (Ph.D.) in the Graduate Program in Molecular and Developmental Biology of the College of Medicine 2001 by Arthur M. Barrie, III B.S., The Ohio State University, 1995 Committee chair: Dr. Bruce Aronow, Ph.D. Abstract Apolipoprotein J (apoJ) is a secreted protein with chaperone homology found in vertebrates and is induced and deposited in both physiologic and pathologic states. ApoJ is hypothesized to be a novel extracellular heat shock protein. I characterized the stress response regulation of the mouse apoJ gene. Cell culture experiments verified the essential roles of an apoJ promoter heat shock element (HSE) and the heat shock transcription factor 1 (HSF1) in heat shock-dependent apoJ gene activation. My studies identified multiple cooperating cis-elements, including a promoter activator protein-1 (AP-1) binding motif, an intron 1 enhancer, and a downstream silencer, that together conferred the full range of apoJ gene expression. -

Drosophila STING Protein Has a Role in Lipid Metabolism
RESEARCH ARTICLE Drosophila STING protein has a role in lipid metabolism Katarina Akhmetova, Maxim Balasov, Igor Chesnokov* Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, School of Medicine, Birmingham, United States Abstract Stimulator of interferon genes (STING) plays an important role in innate immunity by controlling type I interferon response against invaded pathogens. In this work, we describe a previ- ously unknown role of STING in lipid metabolism in Drosophila. Flies with STING deletion are sensi- tive to starvation and oxidative stress, have reduced lipid storage, and downregulated expression of lipid metabolism genes. We found that Drosophila STING interacts with lipid synthesizing enzymes acetyl- CoA carboxylase (ACC) and fatty acid synthase (FASN). ACC and FASN also interact with each other, indicating that all three proteins may be components of a large multi- enzyme complex. The deletion of Drosophila STING leads to disturbed ACC localization and decreased FASN enzyme activity. Together, our results demonstrate a previously undescribed role of STING in lipid metabo- lism in Drosophila. Introduction STimulator of INterferon Genes (STING) is an endoplasmic reticulum (ER)- associated transmembrane protein that plays an important role in innate immune response by controlling the transcription of many host defense genes (Ishikawa and Barber, 2008; Ishikawa et al., 2009; Sun et al., 2009; Tanaka and Chen, 2012; Zhong et al., 2008). The presence of foreign DNA in the cytoplasm signals a danger for the cell. This DNA is recognized by specialized enzyme, the cyclic GMP- AMP synthase (cGAS), which *For correspondence: generates cyclic dinucleotide (CDN) signaling molecules (Diner et al., 2013Li et al., 2013; Gao et al., ichesnokov@ uab. -

Tsukushin LRRC54 Antibody HRP Conjugated
Product Datasheet Tsukushin LRRC54 Antibody HRP Conjugated Catalog No: #C01339H Orders: [email protected] Description Support: [email protected] Product Name Tsukushin LRRC54 Antibody HRP Conjugated Host Species Rabbit Clonality Polyclonal Isotype IgG Purification Purified by Protein A. Applications WB IHC-P Species Reactivity Hu Ms Rt Immunogen Description KLH conjugated synthetic peptide derived from human Tsukushin LRRC54 Conjugates HRP Target Name TSK Tsukushin LRRC54 Other Names E2 induced gene 4 protein; E2IG4; Leucine rich repeat containing protein 54; LRRC54; TSK; Tsukushi; TSK_HUMAN. Concentration 1mg ml Formulation 10mM Tris Buffered Saline containing 1% BSA, 50% glycerol and 0.09% Gentamicin. Storage Store at 4C for 12 months. Application Details Western blotting: 1:100-1000Immunohistochemistry1:100-500 Background The leucine-rich (LRR) repeat is a 20-30 amino acid motif that forms a hydrophobic ? a?? horseshoe fold, allowing it to accommodate several leucine residues within a tightly packed core. All LRR repeats contain a variable segment and a highly conserved segment, the latter of which accounts for 11 or 12 residues of the entire LRR motif. LRRC54 (leucine-rich repeat-containing protein 54), also known as tsukushin, TSKU or E2-induced gene 4 protein (E2IG4), is a 353 amino acid secreted protein that likely localizes to the cell membrane and extracellular compartments. Involved in extracellular secretion and intracellular transport, LRRC54 can be induced by 17-beta-estradiol. Containing nine LRR repeat and a cleavable signal peptide, the gene encoding LRRC54 maps to human chromosome 11, which houses over 1,400 genes and comprises nearly 4% of the human genome. Jervell and Lange-Nielsen syndrome, Jacobsen syndrome, Niemann-Pick disease, hereditary angioedema and Smith-Lemli-Opitz syndrome are associated with defects in genes that maps to chromosome 11. -

Supporting Information SI Materials and Methods Mice And
Supporting Information SI Materials and Methods Mice and Treatments. C57BL/6J mice and ROSA26-CreERT2 mice were purchased from the Jackson Laboratory, and Alk1floxed/floxed mice (1) were transferred to KAIST. To knock down Alk1 in a tamoxifen-dependent manner, Alk12fl/2fl mice were intercrossed with ROSA26-CreERT2 mice. Tamoxifen (0.2 mg; Sigma-Aldrich) dissolved in corn oil (Sigma-Aldrich) was injected into the peritoneal cavity of mouse pups on postnatal day 4 (P4) and P6. All animals were bred in a specific pathogen-free animal facility, and were fed a standard diet (PMI Lab Diet) ad libitum with free access to water. Bmp9 KO mice were generated as previously reported (2). All experimental protocols were performed in accordance with the policies of the Animal Ethics Committee of the University of Tokyo and KAIST. Model of Chronic Aseptic Peritonitis. We used 5-week-old Balb/c mice obtained from Sankyo Laboratories. The model of chronic aseptic peritonitis has been described previously (3-5). To induce peritonitis, we intraperitoneally administered 2 ml of 3% thioglycollate medium (BBL thioglycollate medium, BD Biosciences) to Balb/c mice every 2 days for 2 weeks. The adenovirus encoding lacZ or BMP9 was also intraperitoneally administered twice per week during the same period. Mice were then sacrificed, and their diaphragms were excised and prepared for immunostaining as described previously (3). Fluorescent signals were visualized, and digital images were obtained using a Zeiss LSM 510 confocal microscope equipped with argon and helium-neon lasers (Carl Zeiss). LYVE-1-positive lymphatic vessels were observed using a 20× objective lens and analyzed using LSM Image Browser (Carl Zeiss) and ImageJ software. -

Two Bioactive Molecular Weight Fractions of a Conditioned Medium Enhance RPE Cell Survival on Age-Related Macular Degeneration and Aged Bruch’S Membrane
DOI: 10.1167/tvst.5.1.8 Article Two Bioactive Molecular Weight Fractions of a Conditioned Medium Enhance RPE Cell Survival on Age-Related Macular Degeneration and Aged Bruch’s Membrane Ilene K. Sugino1, Qian Sun1, Carola Springer1, Noounanong Cheewatrakoolpong1, Tong Liu2, Hong Li2, and Marco A. Zarbin1 1 Institute of Ophthalmology and Visual Science, Rutgers, New Jersey Medical School, Newark, NJ, USA 2 Department of Biochemistry and Molecular Biology, Center for Advanced Proteomics Research, Neuroproteomics Core Facility, Rutgers, New Jersey Medical School, Newark, NJ, USA Correspondence: Marco A. Zarbin, Purpose: To characterize molecular weight fractions of bovine corneal endothelial cell Institute of Ophthalmology and conditioned medium (CM) supporting retinal pigment epithelium (RPE) cell survival Visual Science, 90 Bergen Street, on aged and age-related macular degeneration (AMD) Bruch’s membrane. Newark, NJ 07101-1709, USA; [email protected] Methods: CM was subject to size separation using centrifugal filters. Retentate and filtrate fractions were tested for bioactivity by analyzing RPE survival on submacular Received: 4 September 2015 Bruch’s membrane of aged and AMD donor eyes and behavior on collagen I-coated Accepted: 1 January 2016 tissue culture wells. Protein and peptide composition of active fractions was Published: 22 February 2016 determined by mass spectrometry. Keywords: age-related macular Results: Two bioactive fractions, 3-kDa filtrate and a 10-50–kDa fraction, were degeneration; retinal pigment ep- necessary for RPE survival on aged and AMD Bruch’s membrane. The 3-kDa filtrate, ithelium; cell transplantation; but not the 10-50–kDa fraction, supported RPE growth on collagen 1-coated tissue Bruch’s membrane; cell survival culture plates. -

Transcriptional Profiling of Inductive Mesenchyme to Identify Molecules Involved in Prostate Development and Disease
Open Access Research2007VanpouckeetVolume al. 8, Issue 10, Article R213 Transcriptional profiling of inductive mesenchyme to identify molecules involved in prostate development and disease Griet Vanpoucke¤*, Brigid Orr¤*, O Cathal Grace*, Ray Chan*, George R Ashley*, Karin Williams†, Omar E Franco†, Simon W Hayward† and Axel A Thomson* Addresses: *MRC Human Reproductive Sciences Unit, The Queens Medical Research Institute, Little France Crescent, Edinburgh EH16 4TJ, UK. †Departments of Urologic Surgery and Cancer Biology, Vanderbilt University Medical Center, 21st Avenue South, Nashville, TN 37232- 2765, USA. ¤ These authors contributed equally to this work. Correspondence: Axel A Thomson. Email: [email protected] Published: 8 October 2007 Received: 30 March 2007 Revised: 31 May 2007 Genome Biology 2007, 8:R213 (doi:10.1186/gb-2007-8-10-r213) Accepted: 8 October 2007 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2007/8/10/R213 © 2007 Vanpoucke et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Expression<p>Comparisonwereinvolved enriched in prostateprofiling in, of or SAGE developmentspecificprostatic libraries to, inductive inductive and for prostatictumorigenesis.</p> mesenchyme mesenchyme. inductive Further mesenchyme analysis and suggested the complete that <it>Scube1 prostatic rudiment </it>is a revealednovel stromal 219 transcripts molecule that Abstract Background: The mesenchymal compartment plays a key role in organogenesis, and cells within the mesenchyme/stroma are a source of potent molecules that control epithelia during development and tumorigenesis. -

Early-Life Social Experience Affects Offspring DNA Methylation and Later Life Stress Phenotype ✉ Zachary M
ARTICLE https://doi.org/10.1038/s41467-021-24583-x OPEN Early-life social experience affects offspring DNA methylation and later life stress phenotype ✉ Zachary M. Laubach 1,2,3,4,13 , Julia R. Greenberg1,2,4, Julie W. Turner 1,2,3,4, Tracy M. Montgomery1,2,4,5, Malit O. Pioon4, Maggie A. Sawdy1,2, Laura Smale1,6, Raymond G. Cavalcante 7, Karthik R. Padmanabhan7, Claudia Lalancette7, Bridgett vonHoldt 8, Christopher D. Faulk 9, Dana C. Dolinoy7,10,11, Kay E. Holekamp 1,2,3,4 & Wei Perng12 1234567890():,; Studies in rodents and captive primates suggest that the early-life social environment affects future phenotype, potentially through alterations to DNA methylation. Little is known of these associations in wild animals. In a wild population of spotted hyenas, we test the hypothesis that maternal care during the first year of life and social connectedness during two periods of early development leads to differences in DNA methylation and fecal glucocorticoid meta- bolites (fGCMs) later in life. Here we report that although maternal care and social con- nectedness during the den-dependent life stage are not associated with fGCMs, greater social connectedness during the subadult den-independent life stage is associated with lower adult fGCMs. Additionally, more maternal care and social connectedness after den inde- pendence correspond with higher global (%CCGG) DNA methylation. We also note differ- ential DNA methylation near 5 genes involved in inflammation, immune response, and aging that may link maternal care with stress phenotype. 1 Department of Integrative Biology, Michigan State University, East Lansing, MI, USA. 2 Program in Ecology, Evolution, and Behavior, Michigan State University, East Lansing, USA MI, USA. -
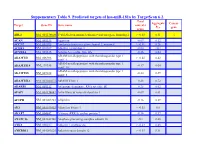
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Ruth Fuhrman-Luck Thesis (PDF 6MB)
DETERMINING THE KALLIKREIN- RELATED PEPTIDASE 4-REGULATED TRANSCRIPTOME AND DEGRADOME IN THE PROSTATE TUMOUR MICROENVIRONMENT Ruth Anna Fuhrman-Luck BAppSc(Hons) Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Institute of Health and Biomedical Innovation Faculty of Health School of Biomedical Sciences Queensland University of Technology May 2015 Keywords Cancer, degradome, degradomics, fibroblast, kallikrein-related peptidase, microenvironment, myofibroblast, omics, peptidase, prostate, protease, proteomics, reactive, stroma, substrate, transcriptome, transcriptomics, transforming growth factor, tumour. i ii Abstract Prostate cancer is the third-leading cause of male cancer-related deaths in developed countries. Prostate cancer-associated mortality ensues when the cancer spreads to other organs, primarily bone. Novel therapies are required to inhibit prostate cancer development and progression to malignant disease. While it is the gland-lining or epithelial cells of the prostate that become malignant, these cells must activate the surrounding microenvironment to facilitate disease progression. As such, future anti- cancer therapies should target factors that drive development of the tumour and activation of its microenvironment. Kallikrein-related peptidase 4 (KLK4) is a prostate epithelial cell-secreted protease, which is over-produced in prostate cancer and in a pre-cancerous pathology whereupon activation of the tumour microenvironment is believed to initiate. The pattern of expression of KLK4 in prostate pathologies suggests that it may function in prostate cancer progression. A small number of functional studies performed to date have shown that KLK4 induces prostate cancer cell motility and transformation to a migratory phenotype, each of which precedes the spread of localised disease and metastasis. However, knowledge of KLK4 function in the surrounding tumour microenvironment is limited.