Truncating Variant Burden in High Functioning Autism and Pleiotropic Effects of LRP1 Across Psychiatric Phenotypes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reconfiguring Nature's Cholesterol Accepting Lipoproteins As
nanomaterials Review Reconfiguring Nature’s Cholesterol Accepting Lipoproteins as Nanoparticle Platforms for Transport and Delivery of Therapeutic and Imaging Agents , Skylar T. Chuang y z, Siobanth Cruz y and Vasanthy Narayanaswami * Department of Chemistry and Biochemistry, California State University, Long Beach, 1250 Bellflower Blvd, Long Beach, CA 90840, USA; [email protected] (S.T.C.); [email protected] (S.C.) * Correspondence: [email protected]; Tel.: +1-562-985-4953; Fax: +1-562-985-8557 These authors contributed equally to this work. y Current address: Department of Chemistry and Chemical Biology, Rutgers University, 123 Bevier Road, z Piscataway, NJ 08854, USA. Received: 2 March 2020; Accepted: 29 April 2020; Published: 8 May 2020 Abstract: Apolipoproteins are critical structural and functional components of lipoproteins, which are large supramolecular assemblies composed predominantly of lipids and proteins, and other biomolecules such as nucleic acids. A signature feature of apolipoproteins is the preponderance of amphipathic α-helical motifs that dictate their ability to make extensive non-covalent inter- or intra-molecular helix–helix interactions in lipid-free states or helix–lipid interactions with hydrophobic biomolecules in lipid-associated states. This review focuses on the latter ability of apolipoproteins, which has been capitalized on to reconstitute synthetic nanoscale binary/ternary lipoprotein complexes composed of apolipoproteins/peptides and lipids that mimic native high-density lipoproteins (HDLs) with the goal to transport drugs. It traces the historical development of our understanding of these nanostructures and how the cholesterol accepting property of HDL has been reconfigured to develop them as drug-loading platforms. The review provides the structural perspective of these platforms with different types of apolipoproteins and an overview of their synthesis. -

Sleeping Beauty Transposon Mutagenesis Identifies Genes That
Sleeping Beauty transposon mutagenesis identifies PNAS PLUS genes that cooperate with mutant Smad4 in gastric cancer development Haruna Takedaa,b, Alistair G. Rustc,d, Jerrold M. Warda, Christopher Chin Kuan Yewa, Nancy A. Jenkinsa,e, and Neal G. Copelanda,e,1 aDivision of Genomics and Genetics, Institute of Molecular and Cell Biology, Agency for Science, Technology and Research, Singapore 138673; bDepartment of Pathology, School of Medicine, Kanazawa Medical University, Ishikawa 920-0293, Japan; cExperimental Cancer Genetics, Wellcome Trust Sanger Institute, Cambridge CB10 1HH, United Kingdom; dTumour Profiling Unit, The Institute of Cancer Research, Chester Beatty Laboratories, London SW3 6JB, United Kingdom; and eCancer Research Program, Houston Methodist Research Institute, Houston, TX 77030 Contributed by Neal G. Copeland, February 27, 2016 (sent for review October 15, 2015; reviewed by Yoshiaki Ito and David A. Largaespada) Mutations in SMAD4 predispose to the development of gastroin- animal models that mimic human GC, researchers have infected testinal cancer, which is the third leading cause of cancer-related mice with H. pylori and then, treated them with carcinogens. They deaths. To identify genes driving gastric cancer (GC) development, have also used genetic engineering to develop a variety of trans- we performed a Sleeping Beauty (SB) transposon mutagenesis genic and KO mouse models of GC (10). Smad4 KO mice are one + − screen in the stomach of Smad4 / mutant mice. This screen iden- GC model that has been of particular interest to us (11, 12). tified 59 candidate GC trunk drivers and a much larger number of Heterozygous Smad4 KO mice develop polyps in the pyloric re- candidate GC progression genes. -

LRP1B Mutations Are Associated with Favorable Outcomes to Immune Checkpoint Inhibitors Across Multiple Cancer Types
Open access Original research J Immunother Cancer: first published as 10.1136/jitc-2020-001792 on 2 March 2021. Downloaded from LRP1B mutations are associated with favorable outcomes to immune checkpoint inhibitors across multiple cancer types 1 2 3 4 Landon C Brown , Matthew D Tucker , Ramy Sedhom, Eric B Schwartz, 5 1 1 1 Jason Zhu, Chester Kao, Matthew K Labriola , Rajan T Gupta, 1 6 1 1 1 Daniele Marin, Yuan Wu, Santosh Gupta, Tian Zhang , Michael R Harrison, Daniel J George,1 Ajjai Alva,4 Emmanuel S Antonarakis,3 Andrew J Armstrong1 To cite: Brown LC, Tucker MD, ABSTRACT lung cancer (NSCLC) and renal cell carci- Sedhom R, et al. LRP1B Background Low- density lipoprotein receptor- related noma.1–3 However, not all patients respond mutations are associated with protein 1b (encoded by LRP1B) is a putative tumor favorable outcomes to immune to treatment with ICIs. The use of predictive suppressor, and preliminary evidence suggests LRP1B- checkpoint inhibitors across biomarkers for response has been explored mutated cancers may have improved outcomes with multiple cancer types. Journal in many tumor types with varying degrees immune checkpoint inhibitors (ICI). for ImmunoTherapy of Cancer of success. Programmed-death ligand-1 (PD- Methods We conducted a multicenter, retrospective pan- 2021;9:e001792. doi:10.1136/ L1) expression,1 microsatellite instability jitc-2020-001792 cancer analysis of patients with LRP1B alterations treated 4 with ICI at Duke University, Johns Hopkins University (JHU) (MSI)/mismatch repair deficiency and 5 6 These data were presented at and University of Michigan (UM). The primary objective tumor mutational burden (TMB) are the the ASCO annual meeting 2020, was to assess the association between overall response only FDA- approved predictive biomarkers for Abstract # 3007. -

Identification of LRP1B-Interacting Proteins and Inhibition of Protein
FEBS Letters 583 (2009) 43–48 journal homepage: www.FEBSLetters.org Identification of LRP1B-interacting proteins and inhibition of protein kinase Ca-phosphorylation of LRP1B by association with PICK1 Tomoko Shiroshima, Chio Oka, Masashi Kawaichi * Division of Gene Function in Animals, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan article info abstract Article history: Recent studies show LDL receptor-related protein 1B, LRP1B as a transducer of extracellular signals. Received 17 October 2008 Here, we identify six interacting partners of the LRP1B cytoplasmic region by yeast two-hybrid Revised 9 November 2008 screen and confirmed their in vivo binding by immunoprecipitation. One of the partners, PICK1 rec- Accepted 18 November 2008 ognizes the C-terminus of LRP1B and LRP1. The cytoplasmic domains of LRP1B are phosphorylated Available online 9 December 2008 by PKCa about 100 times more efficiently than LRP1. Binding of PICK1 inhibits phosphorylation of Edited by Gianni Cesareni LRP1B, but does not affect LRP1 phosphorylation. This study presents the possibility that LRP1B participates in signal transduction which PICK1 may regulate by inhibiting PKC phosphorylation of LRP1B. Keywords: a LDL receptor family LRP1B Structured summary: LRP1 MINT-6801075: Lrp1b (uniprotkb:Q9JI18) physically interacts (MI:0218) with SNTG2 (uniprotkb:Q925E0) PICK1 by two hybrid (MI:0018) JIP MINT-6801030, MINT-6801468: Lrp1b (uniprotkb:Q9JI18) physically interacts (MI:0218) with Pick1 (uni- PDZ domain protkb:Q80VC8) by two -

LRP1 Regulates Peroxisome Biogenesis and Cholesterol
RESEARCH ARTICLE LRP1 regulates peroxisome biogenesis and cholesterol homeostasis in oligodendrocytes and is required for proper CNS myelin development and repair Jing-Ping Lin1, Yevgeniya A Mironova2, Peter Shrager3, Roman J Giger1,2,4,5* 1Department of Cell and Developmental Biology, University of Michigan School of Medicine, Ann Arbor, MI, United States; 2Cellular and Molecular Biology Graduate Program, University of Michigan Medical School, Ann Arbor, MI, United States; 3Department of Neuroscience, University of Rochester Medical Center, Rochester, NY, United States ; 4Department of Neurology, University of Michigan Medical School, Ann Arbor, MI, United States; 5Interdepartmental Neuroscience Graduate Program, University of Michigan Medical School, Ann Arbor, MI, United States Abstract Low-density lipoprotein receptor-related protein-1 (LRP1) is a large endocytic and signaling molecule broadly expressed by neurons and glia. In adult mice, global inducible (Lrp1flox/ flox;CAG-CreER) or oligodendrocyte (OL)-lineage specific ablation (Lrp1flox/flox;Pdgfra-CreER) of Lrp1 attenuates repair of damaged white matter. In oligodendrocyte progenitor cells (OPCs), Lrp1 is required for cholesterol homeostasis and differentiation into mature OLs. Lrp1-deficient OPC/ OLs show a strong increase in the sterol-regulatory element-binding protein-2 yet are unable to maintain normal cholesterol levels, suggesting more global metabolic deficits. Mechanistic studies revealed a decrease in peroxisomal biogenesis factor-2 and fewer peroxisomes in OL processes. / *For correspondence: Treatment of Lrp1À À OPCs with cholesterol or activation of peroxisome proliferator-activated [email protected] receptor-g with pioglitazone alone is not sufficient to promote differentiation; however, when combined, cholesterol and pioglitazone enhance OPC differentiation into mature OLs. Collectively, Competing interests: The our studies reveal a novel role for Lrp1 in peroxisome biogenesis, lipid homeostasis, and OPC authors declare that no competing interests exist. -

A New Role for LRP2 in Forebrain Development
A new role for LRP2 in forebrain development DISSERTATION zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) im Fach Biologie eingereicht an der Mathematisch-Naturwissenschaftlichen Fakult¨atI Humboldt-Universit¨atzu Berlin von Herr Dipl.-Biol. (technisch orientiert) Uwe Anzenberger geboren am 22.03.1976 in Singen a. Htwl. Pr¨asident der Humboldt-Universit¨atzu Berlin: Prof. Dr. Christoph Markschies Dekan der Mathematisch-Naturwissenschaftlichen Fakult¨atI: Prof. Thomas Buckhout, Phd Gutachter: 1. Prof. Dr. Thomas Willnow 2. Prof. Dr. Michael Bader 3. Prof. Dr. Wolfgang Lockau Tag der m¨undlichen Pr¨ufung: 22. September 2006 Abstract LRP2 is a member of the low-density lipoprotein receptor gene family that is mainly expressed in the yolk sac and in the neuroepithelium of the early embryo. Deficiency for this 600 kDa protein in mice results in holoprosencephaly, indicating an important yet unknown role for LRP2 in forebrain development. In this study, mice with a complete or a conditional loss of lrp2 function were used to further elucidate the consequences of the lack of LRP2 expression. This study shows that the presence of LRP2 in the neuroepithelium but not in the yolk sac is crucial for early forebrain development. Lack of the receptor resulted in an increase of Bone morphogenic protein (Bmp) 4 signaling in the rostral telencephalon at E9.5. As a consequence, sonic hedgehog (shh) expression at E10.5 was lost completely in a ventral region of the telencephalon termed anterior entopeduncular area (AEP). The absence of Shh activity in this area subsequently led to the loss of ventrally induced oligodendroglial and interneuronal cell populations in lrp2 deficient mice. -

Convergence of Genes Implicated in Alzheimer's Disease on the Cerebral
Neurochemistry International 50 (2007) 12–38 www.elsevier.com/locate/neuint Review Convergence of genes implicated in Alzheimer’s disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis C.J. Carter 176 Downs Road, Hastings, East Sussex TN34 2DZ, UK Received 5 April 2006; received in revised form 30 June 2006; accepted 11 July 2006 Available online 12 September 2006 Abstract Polymorphic genes associated with Alzheimer’s disease (see www.polygenicpathways.co.uk) delineate a clearly defined pathway related to cerebral and peripheral cholesterol and lipoprotein homoeostasis. They include all of the key components of a glia/neurone cholesterol shuttle including cholesterol binding lipoproteins APOA1, APOA4, APOC1, APOC2, APOC3, APOD, APOE and LPA, cholesterol transporters ABCA1, ABCA2, lipoprotein receptors LDLR, LRP1, LRP8 and VLDLR, and the cholesterol metabolising enzymes CYP46A1 and CH25H, whose oxysterol products activate the liver X receptor NR1H2 and are metabolised to esters by SOAT1. LIPA metabolises cholesterol esters, which are transported by the cholesteryl ester transport protein CETP. The transcription factor SREBF1 controls the expression of most enzymes of cholesterol synthesis. APP is involved in this shuttle as it metabolises cholesterol to 7-betahydroxycholesterol, a substrate of SOAT1 and HSD11B1, binds to APOE and is tethered to LRP1 via APPB1, APBB2 and APBB3 at the cytoplasmic domain and via LRPAP1 at the extracellular domain. APP cleavage products are also able to prevent cholesterol binding to APOE. BACE cleaves both APP and LRP1. Gamma-secretase (PSEN1, PSEN2, NCSTN) cleaves LRP1 and LRP8 as well as APP and their degradation products control transcription factor TFCP2, which regulates thymidylate synthase (TS) and GSK3B expression. -

Disruption of Nucleocytoplasmic Trafficking As a Cellular Senescence
www.nature.com/emm ARTICLE OPEN Disruption of nucleocytoplasmic trafficking as a cellular senescence driver Ji-Hwan Park1,14, Sung Jin Ryu2,13,14, Byung Ju Kim3,13,14, Hyun-Ji Cho3, Chi Hyun Park4, Hyo Jei Claudia Choi2, Eun-Jin Jang3, Eun Jae Yang5, Jeong-A Hwang5, Seung-Hwa Woo5, Jun Hyung Lee5, Ji Hwan Park5, Kyung-Mi Choi6, Young-Yon Kwon6, 6 7 3 3 8 9 10 5 ✉ Cheol-Koo Lee , Joon✉ Tae Park , Sung✉ Chun Cho , Yun-Il Lee , Sung✉ Bae Lee , Jeong A. Han , Kyung A. Cho , Min-Sik Kim , Daehee Hwang11 , Young-Sam Lee3,5 and Sang Chul Park3,12 © The Author(s) 2021 Senescent cells exhibit a reduced response to intrinsic and extrinsic stimuli. This diminished reaction may be explained by the disrupted transmission of nuclear signals. However, this hypothesis requires more evidence before it can be accepted as a mechanism of cellular senescence. A proteomic analysis of the cytoplasmic and nuclear fractions obtained from young and senescent cells revealed disruption of nucleocytoplasmic trafficking (NCT) as an essential feature of replicative senescence (RS) at the global level. Blocking NCT either chemically or genetically induced the acquisition of an RS-like senescence phenotype, named nuclear barrier-induced senescence (NBIS). A transcriptome analysis revealed that, among various types of cellular senescence, NBIS exhibited a gene expression pattern most similar to that of RS. Core proteomic and transcriptomic patterns common to both RS and NBIS included upregulation of the endocytosis-lysosome network and downregulation of NCT in senescent cells, patterns also observed in an aging yeast model. -

Prognostic Analysis of Tumor Mutation Burden and Immune Infiltration in Hepatocellular Carcinoma Based on TCGA Data
www.aging-us.com AGING 2021, Vol. 13, No. 8 Research Paper Prognostic analysis of tumor mutation burden and immune infiltration in hepatocellular carcinoma based on TCGA data Shaoqing Liu1,2,*, Qianjie Tang3,*, Jianwen Huang2,*, Meixiao Zhan2, Wei Zhao2, Xiangyu Yang2, Yong Li2, Lige Qiu2, Fujun Zhang1, Ligong Lu2,&, Xu He2 1Department of Minimally Invasive and Interventional Radiology, Sun Yat-Sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou 510060, China 2Zhuhai Interventional Medical Center, Zhuhai Precision Medical Center, Zhuhai People's Hospital Affiliated with Jinan University, Jinan University, Zhuhai 519000, China 3Department of Pharmacy, Guangdong Women and Children Hospital, Guangzhou 511400, China *Equal contribution Correspondence to: Fujun Zhang, Ligong Lu, Xu He; email: [email protected], [email protected], https://orcid.org/0000-0002-7850-7640; [email protected], https://orcid.org/0000-0002-7565-5111 Keywords: hepatocellular carcinoma, tumor mutation burden, The Cancer Genome Atlas, immune infiltration Received: September 24, 2020 Accepted: January 14, 2021 Published: April 4, 2021 Copyright: © 2021 Liu et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT In order to explore the prognosis of tumor mutation burden (TMB) and the relationship with tumor infiltrating immune cells in hepatocellular carcinoma (HCC), we downloaded somatic mutation data and transcriptome profiles of 376 HCC patients from The Cancer Genome Atlas (TCGA) cohort. We divided the samples into high- TMB and low-TMB groups. -

Gene List HTG Edgeseq Oncology Biomarker Panel
Gene List HTG EdgeSeq Oncology Biomarker Panel For Research Use Only. Not for use in diagnostic procedures. A2M ADRA2B APH1B BAG1 BRCA2 CARM1 CCNH CDC25A CHI3L1 COX7B CXCL16 DESI1 ABCA2 ADRA2C APOC2 BAG2 BRIP1 CASP1 CCNO CDC25B CHI3L2 CP CXCL2 DFFA ABCA3 AFF1 APOC4 BAG3 BTC CASP10 CCNT1 CDC25C CHMP4B CPT1A CXCL3 DHCR24 ABCA4 AGER APOL3 BAG4 BTG1 CASP12 CCR1 CDC34 CHPT1 CPT1B CXCL5 DHH ABCA5 AGFG1 APP BAG5 BTG2 CASP14 CCR10 CDC42 CHRNA1 CPT1C CXCL6 DHX58 ABCA9 AGGF1 APPBP2 BAI1 BTG3 CASP2 CCR2 CDC42BPA CHRNB1 CPT2 CXCL8 DIABLO ABCB11 AGT AQP1 BAIAP3 BTK CASP3 CCR3 CDC6 CHSY1 CRADD CXCL9 DIAPH3 ABCB4 AHNAK AQP2 BAK1 BTRC CASP4 CCR4 CDC7 CHUK CREB1 CXCR1 DICER1 ABCB5 AHNAK2 AQP4 BAMBI BUB1 CASP5 CCR5 CDCA7 CIC CREB3L1 CXCR2 DISP1 ABCB6 AHR AQP7 BAP1 BUB1B CASP6 CCR6 CDH1 CIDEA CREB3L3 CXCR3 DISP2 ABCC1 AHRR AQP9 BATF C17orf53 CASP7 CCR7 CDH13 CIDEB CREB3L4 CXCR4 DKC1 ABCC10 AICDA AR BAX C19orf40 CASP8 CCR8 CDH15 CIRBP CREB5 CXCR5 DKK1 ABCC11 AIFM1 ARAF BBC3 C1orf106 CASP8AP2 CCR9 CDH2 CITED2 CREBBP CXCR6 DKK2 ABCC12 AIMP2 AREG BBS4 C1orf159 CASP9 CCRL2 CDH3 CKB CRK CXXC4 DKK3 ABCC2 AK1 ARHGAP44 BCAR1 C1orf86 CAV1 CCS CDH5 CKLF CRLF2 CXXC5 DKK4 ABCC3 AK2 ARHGEF16 BCAT1 C1QA CAV2 CCT2 CDK1 CKMT1A CRLS1 CYBA DLC1 ABCC4 AK3 ARID1A BCCIP C1S CBL CCT3 CDK16 CKMT2 CRP CYBB DLGAP5 ABCC5 AKAP1 ARID1B BCL10 C3 CBLC CCT4 CDK2 CKS1B CRTAC1 CYCS DLK1 ABCC6 AKR1B1 ARID2 BCL2 C3AR1 CBX3 CCT5 CDK4 CKS2 CRTC2 CYLD DLL1 ABCD1 AKR1C3 ARMC1 BCL2A1 C5 CBX5 CCT6A CDK5 CLCA2 CRY1 CYP19A1 DLL3 ABCD3 AKT1 ARNT BCL2L1 C5AR1 CCBL2 CCT6B CDK5R1 CLCF1 CRYAA CYP1A1 DLL4 -
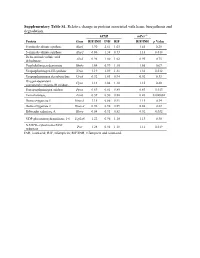
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

LRP1B Deletion in High-Grade Serous Ovarian Cancers Is Associated with Acquired Chemotherapy Resistance to Liposomal Doxorubicin
Cancer Molecular and Cellular Pathobiology Research LRP1B Deletion in High-Grade Serous Ovarian Cancers Is Associated with Acquired Chemotherapy Resistance to Liposomal Doxorubicin Prue A. Cowin1,3,4, Joshy George1,5, Sian Fereday1, Elizabeth Loehrer1, Peter Van Loo6,7, Carleen Cullinane1,4, Dariush Etemadmoghadam1,4, Sarah Ftouni1, Laura Galletta1, Michael S. Anglesio8, Joy Hendley1, Leanne Bowes1, Karen E. Sheppard2,5, Elizabeth L. Christie1, Australian Ovarian Cancer Study1,9,11, Richard B. Pearson2,3,5, Paul R. Harnett10, Viola Heinzelmann-Schwarz12, Michael Friedlander13, Orla McNally14, Michael Quinn14, Peter Campbell6, Anna deFazio9, and David D.L. Bowtell1,3,4,5 Abstract High-grade serous cancer (HGSC), the most common subtype of ovarian cancer, often becomes resistant to chemotherapy, leading to poor patient outcomes. Intratumoral heterogeneity occurs in nearly all solid cancers, including ovarian cancer, contributing to the development of resistance mechanisms. In this study, we examined the spatial and temporal genomic variation in HGSC using high-resolution single-nucleotide polymorphism arrays. Multiple metastatic lesions from individual patients were analyzed along with 22 paired pretreatment and posttreatment samples. We documented regions of differential DNA copy number between multiple tumor biopsies that correlated with altered expression of genes involved in cell polarity and adhesion. In the paired primary and relapse cohort, we observed a greater degree of genomic change in tumors from patients that were initially sensitive to chemotherapy and had longer progression-free interval compared with tumors from patients that were resistant to primary chemotherapy. Notably, deletion or downregulation of the lipid transporter LRP1B emerged as a significant correlate of acquired resistance in our analysis.