Amorphous Systems Investigated by Nuclear Methods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Small, Young Volcanic Deposits Around the Lunar Farside Craters Rosseland, Bolyai, and Roche
44th Lunar and Planetary Science Conference (2013) 2024.pdf SMALL, YOUNG VOLCANIC DEPOSITS AROUND THE LUNAR FARSIDE CRATERS ROSSELAND, BOLYAI, AND ROCHE. J. H. Pasckert1, H. Hiesinger1, and C. H. van der Bogert1. 1Institut für Planetologie, Westfälische Wilhelms-Universität, Wilhelm-Klemm-Str. 10, 48149 Münster, Germany. jhpasckert@uni- muenster.de Introduction: To understand the thermal evolu- mare basalts on the near- and farside. This gives us the tion of the Moon it is essential to investigate the vol- opportunity to investigate the history of small scale canic history of both the lunar near- and farside. While volcanism on the lunar farside. the lunar nearside is dominated by mare volcanism, the farside shows only some isolated mare deposits in the large craters and basins, like the South Pole-Aitken basin or Tsiolkovsky crater [e.g., 1-4]. This big differ- ence in volcanic activity between the near- and farside is of crucial importance for understanding the volcanic evolution of the Moon. The extensive mare volcanism of the lunar nearside has already been studied in great detail by numerous authors [e.g., 4-8] on the basis of Lunar Orbiter and Apollo data. New high-resolution data obtained by the Lunar Reconnaissance Orbiter (LRO) and the SELENE Terrain Camera (TC) now allow us to investigate the lunar farside in great detail. Basaltic volcanism of the lunar nearside was active for almost 3 Ga, lasting from ~3.9-4.0 Ga to ~1.2 Ga before present [5]. In contrast to the nearside, most eruptions of mare deposits on the lunar farside stopped much earlier, ~3.0 Ga ago [9]. -

U.S. Government Printing Office Style Manual, 2008
U.S. Government Printing Offi ce Style Manual An official guide to the form and style of Federal Government printing 2008 PPreliminary-CD.inddreliminary-CD.indd i 33/4/09/4/09 110:18:040:18:04 AAMM Production and Distribution Notes Th is publication was typeset electronically using Helvetica and Minion Pro typefaces. It was printed using vegetable oil-based ink on recycled paper containing 30% post consumer waste. Th e GPO Style Manual will be distributed to libraries in the Federal Depository Library Program. To fi nd a depository library near you, please go to the Federal depository library directory at http://catalog.gpo.gov/fdlpdir/public.jsp. Th e electronic text of this publication is available for public use free of charge at http://www.gpoaccess.gov/stylemanual/index.html. Use of ISBN Prefi x Th is is the offi cial U.S. Government edition of this publication and is herein identifi ed to certify its authenticity. ISBN 978–0–16–081813–4 is for U.S. Government Printing Offi ce offi cial editions only. Th e Superintendent of Documents of the U.S. Government Printing Offi ce requests that any re- printed edition be labeled clearly as a copy of the authentic work, and that a new ISBN be assigned. For sale by the Superintendent of Documents, U.S. Government Printing Office Internet: bookstore.gpo.gov Phone: toll free (866) 512-1800; DC area (202) 512-1800 Fax: (202) 512-2104 Mail: Stop IDCC, Washington, DC 20402-0001 ISBN 978-0-16-081813-4 (CD) II PPreliminary-CD.inddreliminary-CD.indd iiii 33/4/09/4/09 110:18:050:18:05 AAMM THE UNITED STATES GOVERNMENT PRINTING OFFICE STYLE MANUAL IS PUBLISHED UNDER THE DIRECTION AND AUTHORITY OF THE PUBLIC PRINTER OF THE UNITED STATES Robert C. -

Appendix I Lunar and Martian Nomenclature
APPENDIX I LUNAR AND MARTIAN NOMENCLATURE LUNAR AND MARTIAN NOMENCLATURE A large number of names of craters and other features on the Moon and Mars, were accepted by the IAU General Assemblies X (Moscow, 1958), XI (Berkeley, 1961), XII (Hamburg, 1964), XIV (Brighton, 1970), and XV (Sydney, 1973). The names were suggested by the appropriate IAU Commissions (16 and 17). In particular the Lunar names accepted at the XIVth and XVth General Assemblies were recommended by the 'Working Group on Lunar Nomenclature' under the Chairmanship of Dr D. H. Menzel. The Martian names were suggested by the 'Working Group on Martian Nomenclature' under the Chairmanship of Dr G. de Vaucouleurs. At the XVth General Assembly a new 'Working Group on Planetary System Nomenclature' was formed (Chairman: Dr P. M. Millman) comprising various Task Groups, one for each particular subject. For further references see: [AU Trans. X, 259-263, 1960; XIB, 236-238, 1962; Xlffi, 203-204, 1966; xnffi, 99-105, 1968; XIVB, 63, 129, 139, 1971; Space Sci. Rev. 12, 136-186, 1971. Because at the recent General Assemblies some small changes, or corrections, were made, the complete list of Lunar and Martian Topographic Features is published here. Table 1 Lunar Craters Abbe 58S,174E Balboa 19N,83W Abbot 6N,55E Baldet 54S, 151W Abel 34S,85E Balmer 20S,70E Abul Wafa 2N,ll7E Banachiewicz 5N,80E Adams 32S,69E Banting 26N,16E Aitken 17S,173E Barbier 248, 158E AI-Biruni 18N,93E Barnard 30S,86E Alden 24S, lllE Barringer 29S,151W Aldrin I.4N,22.1E Bartels 24N,90W Alekhin 68S,131W Becquerei -

Calendar Year 2017 Enplanements at All Airports (Primary, Non-Primary
Source: ACAIS Calendar Year 2017 Final 11/7/2018 FAA Airports Revenue Enplanements at All Airports RO ST Locid City Airport Name % Change S/L Hub Rank CY 17 CY 17 CY 16 Enplanements Enplanements Ted Stevens Anchorage 56 AL AK ANC Anchorage International P M 2,556,191 2,563,524 -0.29% 119 AL AK FAI Fairbanks Fairbanks International P S 543,839 538,881 0.92% 133 AL AK JNU Juneau Juneau International P N 422,266 420,442 0.43% 202 AL AK BET Bethel Bethel P N 146,652 155,791 -5.87% 210 AL AK KTN Ketchikan Ketchikan International P N 131,144 127,881 2.55% 237 AL AK ENA Kenai Kenai Municipal P N 92,973 92,485 0.53% 249 AL AK SIT Sitka Sitka Rocky Gutierrez P N 83,598 83,404 0.23% 250 AL AK ADQ Kodiak Kodiak P N 83,577 79,682 4.89% 260 AL AK OTZ Kotzebue Ralph Wien Memorial P N 67,951 64,481 5.38% 263 AL AK OME Nome Nome P N 61,651 60,655 1.64% 283 AL AK HOM Homer Homer P N 46,322 44,117 5.00% Wiley Post-Will Rogers 284 AL AK BRW Barrow Memorial P N 45,958 45,300 1.45% 290 AL AK SCC Deadhorse Deadhorse P N 41,849 51,205 -18.27% 304 AL AK AKN King Salmon King Salmon P N 34,800 36,717 -5.22% 312 AL AK DUT Unalaska Unalaska P N 30,080 31,055 -3.14% 316 AL AK DLG Dillingham Dillingham P N 28,895 33,616 -14.04% 336 AL AK PSG Petersburg Petersburg James A Johnson P N 22,915 21,896 4.65% 348 AL AK LHD Anchorage Lake Hood P N 20,689 23,382 -11.52% 356 AL AK MRI Anchorage Merrill Field P N 18,298 19,144 -4.42% 357 AL AK CDV Cordova Merle K (Mudhole) Smith P N 18,280 18,649 -1.98% 367 AL AK UNK Unalakleet Unalakleet P N 16,111 16,948 -4.94% 378 AL AK WRG Wrangell Wrangell P N 13,160 12,569 4.70% 382 AL AK ANI Aniak Aniak P N 12,735 13,688 -6.96% St. -

Lick Observatory Records: Photographs UA.036.Ser.07
http://oac.cdlib.org/findaid/ark:/13030/c81z4932 Online items available Lick Observatory Records: Photographs UA.036.Ser.07 Kate Dundon, Alix Norton, Maureen Carey, Christine Turk, Alex Moore University of California, Santa Cruz 2016 1156 High Street Santa Cruz 95064 [email protected] URL: http://guides.library.ucsc.edu/speccoll Lick Observatory Records: UA.036.Ser.07 1 Photographs UA.036.Ser.07 Contributing Institution: University of California, Santa Cruz Title: Lick Observatory Records: Photographs Creator: Lick Observatory Identifier/Call Number: UA.036.Ser.07 Physical Description: 101.62 Linear Feet127 boxes Date (inclusive): circa 1870-2002 Language of Material: English . https://n2t.net/ark:/38305/f19c6wg4 Conditions Governing Access Collection is open for research. Conditions Governing Use Property rights for this collection reside with the University of California. Literary rights, including copyright, are retained by the creators and their heirs. The publication or use of any work protected by copyright beyond that allowed by fair use for research or educational purposes requires written permission from the copyright owner. Responsibility for obtaining permissions, and for any use rests exclusively with the user. Preferred Citation Lick Observatory Records: Photographs. UA36 Ser.7. Special Collections and Archives, University Library, University of California, Santa Cruz. Alternative Format Available Images from this collection are available through UCSC Library Digital Collections. Historical note These photographs were produced or collected by Lick observatory staff and faculty, as well as UCSC Library personnel. Many of the early photographs of the major instruments and Observatory buildings were taken by Henry E. Matthews, who served as secretary to the Lick Trust during the planning and construction of the Observatory. -

Palaeoenvironment in North-Western Romania During the Last 15,000 Years
Palaeoenvironment in north-western Romania during the last 15,000 years Angelica Feurdean Dedicated to Ovidiu Feurdean Avhandling i Kvartärgeologi Thesis in Quaternary Geology No. 3 Department of Physical Geography and Quaternary Geology, Stockholm University 2004 1 ISBN 2 Palaeoenvironment in north-western Romania during the last 15,000 years by Angelica Feurdean Department of Physical Geography and Quaternary Geology, Stockholm University, SE-106 91 Stockholm This thesis is based on work carried out as Ph.D. stu- Appendix III: Feurdean A. (in press). Holocene forest dent at the Department of Paleontology, Faculty of Biol- dynamics in north-western Romania. The Holocene. ogy and Geology, Babes-Bolyai University, Cluj-Napoca, Romania between Oct. 1998 and Sept. 2002 and later as Appendix IV: Feurdean A. & Bennike O. Late Quater- Ph.D. student in Quaternary Geology at the Department nary palaeocological and paleoclimatological reconstruc- of Physical Geography and Quaternary Geology, tion in the Gutaiului Mountains, NW Romania. Manu- Stockholm University 2002-2004. The thesis consists of script submitted to Journal of Quaternary Science. four papers and a synthesis. The four papers are listed below and presented in Appendices I-IV. Two of the pa- Fieldwork in Romania has been jointly performed with pers have been published (I, II), one is in press (III) and Barbara Wohlfarth and Leif Björkman (Preluca Tiganului, the fourth has been submitted (IV). The thesis summary Steregoiu, Izvoare) and with Bogdan Onac (Creasta presents an account of earlier pollenstratigraphic work Cocosului). I am responsible for the lithostratigraphic done in Romania and the discussion focuses on tree description of all sediment and peat cores, for sub-sam- dynamics during the Lateglacial and Holocene, based pling and laboratory preparation. -

Harvard University Department of Physics Newsletter
Harvard University Department of Physics Newsletter FALL 2014 COVER STORY Probing the Universe’s Earliest Moments FOCUS Early History of the Physics Department FEATURED Quantum Optics Where Physics Meets Biology Condensed Matter Physics NEWS A Reunion Across Generations and Disciplines 42475.indd 1 10/30/14 9:19 AM Detecting this signal is one of the most important goals in cosmology today. A lot of work by a lot of people has led to this point. JOHN KOVAC, HARVARD-SMITHSONIAN CENTER FOR ASTROPHYSICS LEADER OF THE BICEP2 COLLABORATION 42475.indd 2 10/24/14 12:38 PM CONTENTS ON THE COVER: Letter from the former Chair ........................................................................................................ 2 The BICEP2 telescope at Physics Department Highlights 4 twilight, which occurs only ........................................................................................................ twice a year at the South Pole. The MAPO observatory (home of the Keck Array COVER STORY telescope) and the South Pole Probing the Universe’s Earliest Moments ................................................................................. 8 station can be seen in the background. (Steffen Richter, Harvard University) FOCUS ACKNOWLEDGMENTS Early History of the Physics Department ................................................................................ 12 AND CREDITS: Newsletter Committee: FEATURED Professor Melissa Franklin Professor Gerald Holton Quantum Optics ..............................................................................................................................14 -

INIS Clearinghouse Other IAEA P. 0. Box 100 ^ I L F A-1400, Vienna, Austria
Attention Microfiche User, The original document from which this microfiche was made was found to contain some imperfection or imperfections that reduce full comprehension of some of the text despite the good technical quality of the microfiche itself. The imperfections may be: - missing of illegible pages/figures - wrong pagination - poor overall printing quality, etc. We normally refuse to microfiche such a document and request a replacement document (or pages) from the National INIS Centre concerned. However, our experience shows that many months pass before such documents are replaced. Sometimes the Centre is not able to supply a better copy or, in som, cases, the pages that were supposed to be missing correspond to a wrong pagination only* We feel that it is better to proceed with distributing the microfiche made of these documents than to withhold them till the imperfections are removed. If the removals are subsequestly made then replacement microfiche can be issued. In line with this approach then, our specific practice for microfiching documents with imperfections is as follows: 1* A microfiche of an imperfect document will be marked with a special symbol (black circle) on the left of the title. This symbol will appear on all masters and copies of the document (1st fiche and trailer fiches) even if the imperfection is on one fiche of the report only. 2. If imperfection is not too general the reason will "be specified on a sheet such as this, in the space below. 3. The microfiche will be considered as temporary, but sold at the normal price. Replacements, if they can be issued, will be available for purchase at the regular price. -
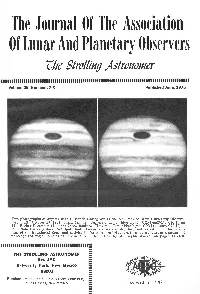
The Journal of the Association of Lunar and Planetary Observers Rite Strolli11g Astro11omer
The Journal Of The Association Of Lunar And Planetary Observers rite Strolli11g Astro11omer 1111111 111111111111111111111111111111111111111111111111111111111111111111111111111111111111111111 Volume 25, Numbers 7-8 Published June, 1975 Two photographs of Jupiter with a 12-inch Cassegrain at the New Mexico State University Observa tory. Left: October 23, 1964; 8hrs., 58mins., Universal Time; blue light; CM(I)=251°, CM(II)= 17° . Right: December 12, 1965; 7hrs., 46mins., Universal Time; blue light; CM(I)=182° , CM(II)= 22° . Note the very dark Red Spot, South Temperate Zone ovals, the great variation in the bright ness of the Equatorial Zone, and activity in far northern belts. Are these events part of a pattern of unrecognized major zenological disturbances? See article by Mr. Wynn Wacker on pages 145-150. 111111111111111111111111111111111111111111111:: THE STROLLING ASTRONOMER - Box lAZ University Park, New Mexico 88003 - Residence telephone 522-4213 (Area Code 505) - in Las Cruces, New Mexico - Founded In 1947 IN THIS ISSUE OBSERVING OLYMPUS MONS IN 1975, by John E. Westfall -------------------------------------------------------------------------- pg. 129 MARS 1973-74 APPARITION- ALPO REPORT I, by Robert B. Rhoads and Virginia W. Capen ------------------------------ pg. 130 A PROPOSED ASTROGEOGRAPHICAL SECTION, by James Powell ---------------------------------------------------------------------·---------- pg. 138 JUPITER IN 1972: ROTATION PERIODS, by Phillip W. Budine ---------------------------------------------------------------------- -

Harold Clayton Urey Papers
http://oac.cdlib.org/findaid/ark:/13030/tf4c6006zq Online items available Harold Clayton Urey Papers Special Collections & Archives, UC San Diego Special Collections & Archives, UC San Diego Copyright 2005 9500 Gilman Drive La Jolla 92093-0175 [email protected] URL: http://libraries.ucsd.edu/collections/sca/index.html Harold Clayton Urey Papers MSS 0044 1 Descriptive Summary Languages: English Contributing Institution: Special Collections & Archives, UC San Diego 9500 Gilman Drive La Jolla 92093-0175 Title: Harold Clayton Urey Papers Identifier/Call Number: MSS 0044 Physical Description: 90.3 Linear feet(157 archives boxes, 34 flat boxes, 5 card file boxes, 1 carton, and 9 art bin items) Date (inclusive): 1924-1981 (bulk 1958-1978) Abstract: Papers of Harold Clayton Urey, Nobel Prize-winning chemist who contributed to significant advances in the fields of physical chemistry, geochemistry, lunar science, and astrochemistry. He received the Nobel Prize for Chemistry in 1934 for his discovery of deuterium, and made key scientific contributions to the development of the atomic bomb during World War II. The papers span the years 1929 to 1981 and contain significant correspondence with Urey's fellow scientists, including Albert Einstein, Enrico Fermi, J. Robert Oppenheimer, Leo Szilard, and Edward Teller. Scope and Content of Collection Papers of Harold Clayton Urey, Nobel Prize-winning chemist who contributed to significant advances in the fields of physical chemistry, geochemistry, lunar science, and astrochemistry. He received the Nobel Prize for Chemistry in 1934 for his discovery of deuterium, and made key scientific contributions to the development of the atomic bomb during World War II. -

Endnotes First Man FSM
Notes Abbreviations Used AB: Alan Bean AC: Andrew Chaikin AF: Arthur Frame ALSJ: Apollo Lunar Surface Journal ALSK: Alma Lou Shaw-Kuffner ATEOS: At the Edge of Space (Thompson) BA: Buzz Aldrin BAP: Bruce A. Peterson BEC: Bruce E. Clingan BG: Bob Gustafson BJC: Bruce J. Clingan BL: Betty Love CCK: Christopher C. Kraft Jr. CDF: Charles D. Friedlander Jr. CE: Cincinnati Enquirer CKA: Carol Knight Armstrong CP: Cincinnati Post CPD: Cleveland Plain Dealer CSM: Charles S. Mechem CTF: Carrying the Fire (Collins) DAA: Dean Alan Armstrong DAG: Donald A. Gardner DJH: Dora Jane Hamblin DS: Dudley Schuler DSS: Donald S. Stephenson EC: Eugene Cernan EFK: Eugene F. Kranz 1 EMB: Ernest M. Beauchamp FB: Frank Borman FOM: First on the Moon (Gene Farmer and Dora Jane Hamblin) GER: George E. “Ernie” Russell GJM: Gene J. Matranga GLW: Gene L. Waltman GWW: Grace Walker-Wiesmann HAG: Herbert A. Graham HCS: Harold C. Schwan HG: Herschel Gott HSC: Harry S. Combs JAH: June Armstrong Hoffman JBB: John “Bud” Blackford JEL: James E. Lovell JG: John Glenn Jr. JGM: John G. McTigue JM: John Moore JSA: Janet Shearon Armstrong JZ: Jacob Zint KCK: Ken C. Kramer KKS: K. K. “Kotcho” Solacoff L: Life magazine LBJ: Lyndon Baines Johnson LN: Lima News (Ohio) MC: Michael Collins MOT: Milton O. Thompson MSC: Manned Spacecraft Center NAA: Neil Alden Armstrong NK: Ned Keiber NM: Norman Mailer NO: The National Observer 2 NPRC: National Personnel Records Center (St. Louis, MO) NYT: New York Times OBR: Onboard Recorder OFM: Of a Fire on the Moon (Mailer) PFB: Paul F. -

Meteoritical Society
Meteoritical Society http://meteoriticalsociety.org 2013 ANNUAL MEETING INVITATION The 76th annual meeting of the Meteoritical Society will take place in Edmonton, the capital city of Alberta, Canada. The meeting will be held on July 29−August 2 at the Crowne Plaza Chateau Lacombe in Edmonton’s downtown core, within walking distance of the Arts District, shopping, nightlife, restaurants, and coffee shops. Edmonton is the most northerly major city in Canada. Long daylight hours and warm weather are therefore expected during the summer months. Situated on the North Saskatchewan River, Edmonton is close to UNESCO World Heritage Sites such as Jasper and Banff national parks in the Canadian Rocky Mountains, Head-Smashed-In Buffalo Jump, and Dinosaur Provincial Park. Less than an hour from Edmonton is Anaïs Fourny, one of the two students sponsored by the Meteoritical Society to attend the workshop. PHOTO CREDIT: ALLAN TREIMAN Elk Island National Park. In addition to being an opportunity to observe bison, moose, and deer in a natural setting, this dark-sky preserve is also a prime viewing area for the aurora borealis, a light show we may fi eld study of a terrestrial analog for lunar crust formation. The be treated to as the next northern lights activity maximum is Meteoritical Society was a cosponsor, and supported attendance by two approached. The conference banquet will be held on Wednesday, July students, Corey Wall and Anaïs Fourny of the University of British 31, on the picturesque campus of the University of Alberta. Columbia. The workshop, held on July 12–16, 2012, was convened by Dr.