Clinical Study Protocol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) Patent Application Publication (10) Pub. No.: US 2004/0224012 A1 Suvanprakorn Et Al
US 2004O224012A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0224012 A1 Suvanprakorn et al. (43) Pub. Date: Nov. 11, 2004 (54) TOPICAL APPLICATION AND METHODS Related U.S. Application Data FOR ADMINISTRATION OF ACTIVE AGENTS USING LIPOSOME MACRO-BEADS (63) Continuation-in-part of application No. 10/264,205, filed on Oct. 3, 2002. (76) Inventors: Pichit Suvanprakorn, Bangkok (TH); (60) Provisional application No. 60/327,643, filed on Oct. Tanusin Ploysangam, Bangkok (TH); 5, 2001. Lerson Tanasugarn, Bangkok (TH); Suwalee Chandrkrachang, Bangkok Publication Classification (TH); Nardo Zaias, Miami Beach, FL (US) (51) Int. CI.7. A61K 9/127; A61K 9/14 (52) U.S. Cl. ............................................ 424/450; 424/489 Correspondence Address: (57) ABSTRACT Eric G. Masamori 6520 Ridgewood Drive A topical application and methods for administration of Castro Valley, CA 94.552 (US) active agents encapsulated within non-permeable macro beads to enable a wider range of delivery vehicles, to provide longer product shelf-life, to allow multiple active (21) Appl. No.: 10/864,149 agents within the composition, to allow the controlled use of the active agents, to provide protected and designable release features and to provide visual inspection for damage (22) Filed: Jun. 9, 2004 and inconsistency. US 2004/0224012 A1 Nov. 11, 2004 TOPCAL APPLICATION AND METHODS FOR 0006 Various limitations on the shelf-life and use of ADMINISTRATION OF ACTIVE AGENTS USING liposome compounds exist due to the relatively fragile LPOSOME MACRO-BEADS nature of liposomes. Major problems encountered during liposome drug Storage in vesicular Suspension are the chemi CROSS REFERENCE TO OTHER cal alterations of the lipoSome compounds, Such as phos APPLICATIONS pholipids, cholesterols, ceramides, leading to potentially toxic degradation of the products, leakage of the drug from 0001) This application claims the benefit of U.S. -

Understanding Benzodiazephine Use, Abuse, and Detection
Siemens Healthcare Diagnostics, the leading clinical diagnostics company, is committed to providing clinicians with the vital information they need for the accurate diagnosis, treatment and monitoring of patients. Our comprehensive portfolio of performance-driven systems, unmatched menu offering and IT solutions, in conjunction with highly responsive service, is designed to streamline workflow, enhance operational efficiency and support improved patient care. Syva, EMIT, EMIT II, EMIT d.a.u., and all associated marks are trademarks of General Siemens Healthcare Diagnostics Inc. All Drugs other trademarks and brands are the Global Division property of their respective owners. of Abuse Siemens Healthcare Product availability may vary from Diagnostics Inc. country to country and is subject 1717 Deerfield Road to varying regulatory requirements. Deerfield, IL 60015-0778 Please contact your local USA representative for availability. www.siemens.com/diagnostics Siemens Global Headquarters Global Siemens Healthcare Headquarters Siemens AG Understanding Wittelsbacherplatz 2 Siemens AG 80333 Muenchen Healthcare Sector Germany Henkestrasse 127 Benzodiazephine Use, 91052 Erlangen Germany Abuse, and Detection Telephone: +49 9131 84 - 0 www.siemens.com/healthcare www.usa.siemens.com/diagnostics Answers for life. Order No. A91DX-0701526-UC1-4A00 | Printed in USA | © 2009 Siemens Healthcare Diagnostics Inc. Syva has been R1 R2 a leading developer N and manufacturer of AB R3 X N drugs-of-abuse tests R4 for more than 30 years. R2 C Now part of Siemens Healthcare ® Diagnostics, Syva boasts a long and Benzodiazepines have as their basic chemical structure successful track record in drugs-of-abuse a benzene ring fused to a seven-membered diazepine ring. testing, and leads the industry in the All important benzodiazepines contain a 5-aryl substituent ring (ring C) and a 1,4–diazepine ring. -

Tel: 86-2985324244; Fax: 86-2985252580
medRxiv preprint doi: https://doi.org/10.1101/2020.02.27.20028605; this version posted February 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC 4.0 International license . Efficacy and Acceptability Comparisons of Cognitive Behavior Therapy, Drugs, and Their Combination for Panic Disorder in Adults: a Network Meta-analysis Fengjie Gao(M.M.)1,a, Hairong He(M.M.)2,a, Bin Yan(M.M.)2, Jian Yang(M.M.)2, Yajuan Fan(M.D.)1, Binbin Zhao(M.M.)1, Xiaoyan He(M.M.)1, Qingyan Ma(M.M.)1, Baijia Li(M.D.)1, Yuan Gao(M.D.)1, Li Qian(M.D.)1, Zai Yang(M.M.)1, Ce Chen(M.M.)1, Yunchun Chen(M.D.)1, Chengge Gao(M.D.)1, Feng Zhu(M.D.)5, Wei Wang(M.M.)1,*, Xiancang Ma(M.D.)1,3,4,* 1Department of Psychiatry, The First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, 710061 Xi’an, Shaanxi, China. 2Clinical Research Center, The First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, 710061 Xi’an, Shaanxi, China. 3Center for Brain Science, The First Affiliated Hospital of Xi’an Jiaotong University, 277 Yanta West Road, 710061 Xi’an, Shaanxi, China. 4Clinical Research center for Psychiatric Medicine of Shaanxi Province, The First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, 710061 Xi’an, Shaanxi, China. -

Analytical Methods for Determination of Benzodiazepines. a Short Review
Cent. Eur. J. Chem. • 12(10) • 2014 • 994-1007 DOI: 10.2478/s11532-014-0551-1 Central European Journal of Chemistry Analytical methods for determination of benzodiazepines. A short review Review Article Paulina Szatkowska1, Marcin Koba1*, Piotr Kośliński1, Jacek Wandas1, Tomasz Bączek2,3 1Department of Toxicology, Faculty of Pharmacy, Collegium Medicum of Nicolaus Copernicus University, 85-089 Bydgoszcz, Poland 2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Medical University of Gdańsk, 80-416 Gdańsk, Poland 3Institute of Health Sciences, Division of Human Anatomy and Physiology, Pomeranian University of Słupsk, 76-200 Słupsk, Poland Received 16 July 2013; Accepted 6 February 2014 Abstract: Benzodiazepines (BDZs) are generally commonly used as anxiolytic and/or hypnotic drugs as a ligand of the GABAA-benzodiazepine receptor. Moreover, some of benzodiazepines are widely used as an anti-depressive and sedative drugs, and also as anti-epileptic drugs and in some cases can be useful as an adjunct treatment in refractory epilepsies or anti-alcoholic therapy. High-performance liquid chromatography (HPLC) methods, thin-layer chromatography (TLC) methods, gas chromatography (GC) methods, capillary electrophoresis (CE) methods and some of spectrophotometric and spectrofluorometric methods were developed and have been extensively applied to the analysis of number of benzodiazepine derivative drugs (BDZs) providing reliable and accurate results. The available chemical methods for the determination of BDZs in biological materials and pharmaceutical formulations are reviewed in this work. Keywords: Analytical methods • Benzodiazepines • Drugs analysis • Pharmaceutical formulations © Versita Sp. z o.o. 1. Introduction and long). For this reason, an application of these drugs became broader allowing their utility to a larger extent, Benzodiazepines have been first introduced into medical and at the same time, problems related to drug abuse practice in the 60s of the last century. -

1 'New/Designer Benzodiazepines'
1 ‘New/Designer Benzodiazepines’: an analysis of the literature and psychonauts’ trip reports 2 Laura Orsolini*1,2,3, John M. Corkery1, Stefania Chiappini1, Amira Guirguis1, Alessandro Vento4,5,6,7, 3 Domenico De Berardis3,8,9, Duccio Papanti1, and Fabrizio Schifano1 4 5 1 Psychopharmacology, Drug Misuse and Novel Psychoactive Substances Research Unit, School of Life and Medical 6 Sciences, University of Hertfordshire, Hatfield, AL10 9AB, Herts, UK. 7 2 Neomesia Mental Health, Villa Jolanda Hospital, Jesi, Italy. 8 3 Polyedra, Teramo, Italy. 9 4 NESMOS Department (Neurosciences, Mental Health and Sensory Organs), Sapienza University – Rome, School of 10 Medicine and Psychology; Sant’Andrea Hospital, Rome, Italy 11 5 School of psychology - G. Marconi Telematic University, Rome, Italy 12 6 Addictions Observatory (ODDPSS), Rome, Italy 13 7 Mental Health Department - ASL Roma 2, Rome, Italy 14 8 Department of Neuroscience, Imaging and Clinical Science, Chair of Psychiatry, University of “G. D’Annunzio”, Chieti, 15 Italy. 16 9 NHS, Department of Mental Health, Psychiatric Service of Diagnosis and Treatment, Hospital “G. Mazzini”, ASL 4 17 Teramo, Italy. 18 19 Corresponding author: 20 Laura Orsolini, Psychopharmacology, Drug Misuse and Novel Psychoactive Substances Research Unit, School of Life 21 and Medical Sciences, University of Hertfordshire, Hatfield, AL10 9AB, Herts, UK; Villa Jolanda Hospital, Neomesia 22 Mental Health, Villa Jolanda, Italy; Polyedra, Teramo, Italy; E-mail address: [email protected]. Tel.: (+39) 392 23 3244643. 24 25 Conflicts of Interest 26 The authors declare that this research was conducted in the absence of any commercial or financial relationships 27 that could be construed as a potential conflict of interest. -
![Characterization of the Binding and Comparison of the Distribution of Benzodiazepine Receptors Labeled with Ph1diazepam and [3H]Alprazolam L&S K](https://docslib.b-cdn.net/cover/7046/characterization-of-the-binding-and-comparison-of-the-distribution-of-benzodiazepine-receptors-labeled-with-ph1diazepam-and-3h-alprazolam-l-s-k-1187046.webp)
Characterization of the Binding and Comparison of the Distribution of Benzodiazepine Receptors Labeled with Ph1diazepam and [3H]Alprazolam L&S K
fIivIoPSYCHOPHARMACOLOGY 1993-VOL. 8, NO.4 305 Characterization of the Binding and Comparison of the Distribution of Benzodiazepine Receptors Labeled with pH1Diazepam and [3H]Alprazolam l&s K. Wamsley, Ph.D., Lisa L. Longlet, B.S., MaryAnne E. Hunt, Ph.D., Donald R. Mahan, IfiMilrio E. Alburges, Ph.D. Ttrbinding characteristics of [3H]diazepam and benzodiazepine drugs and apparently do not overlap onto �prazolam were obtained by in vitro analysis of other subtypes of receptors. These experiments were IIfiorrsof rat brain. Dissociation, association, and performed by both binding assay in tissue sections and by sDlItionaTUllyses were performed to optimize the light microscopic autoradiography. The major difference GdilWnsfor obtaining selective labeling of between the labeling of the two compounds is· represented llaldiDzepinerecept ors with the two tritiated by the peripheral benzodiazepine sites, which are GIIpOUnds. Both drugs approached equilibrium rapidly recognized by [3H]diazepam, but not occupied by nntro. Rosenthal analysis (Scatchard plot) of the [3H]alprazolam (at nanomolar concentrations). This amiondata indicated a similar finite number of difference was readily apparent in the auto radiograms. IIII;otSwas being occupied by both ligands. Other pharmacokinetic or pharmacodynamic properties Clapetitionstudies, using various ligands to inhibit both must distinguish these two benzodiazepines. fH}bu.tpam and [3H]alprazolam indicated that these [Neuropsychopharmacology 8:305-314, 1993J tIIIcmnpoundsbind to the tissue sections as typical -

Drug and Medication Classification Schedule
KENTUCKY HORSE RACING COMMISSION UNIFORM DRUG, MEDICATION, AND SUBSTANCE CLASSIFICATION SCHEDULE KHRC 8-020-1 (11/2018) Class A drugs, medications, and substances are those (1) that have the highest potential to influence performance in the equine athlete, regardless of their approval by the United States Food and Drug Administration, or (2) that lack approval by the United States Food and Drug Administration but have pharmacologic effects similar to certain Class B drugs, medications, or substances that are approved by the United States Food and Drug Administration. Acecarbromal Bolasterone Cimaterol Divalproex Fluanisone Acetophenazine Boldione Citalopram Dixyrazine Fludiazepam Adinazolam Brimondine Cllibucaine Donepezil Flunitrazepam Alcuronium Bromazepam Clobazam Dopamine Fluopromazine Alfentanil Bromfenac Clocapramine Doxacurium Fluoresone Almotriptan Bromisovalum Clomethiazole Doxapram Fluoxetine Alphaprodine Bromocriptine Clomipramine Doxazosin Flupenthixol Alpidem Bromperidol Clonazepam Doxefazepam Flupirtine Alprazolam Brotizolam Clorazepate Doxepin Flurazepam Alprenolol Bufexamac Clormecaine Droperidol Fluspirilene Althesin Bupivacaine Clostebol Duloxetine Flutoprazepam Aminorex Buprenorphine Clothiapine Eletriptan Fluvoxamine Amisulpride Buspirone Clotiazepam Enalapril Formebolone Amitriptyline Bupropion Cloxazolam Enciprazine Fosinopril Amobarbital Butabartital Clozapine Endorphins Furzabol Amoxapine Butacaine Cobratoxin Enkephalins Galantamine Amperozide Butalbital Cocaine Ephedrine Gallamine Amphetamine Butanilicaine Codeine -

A Review of the Evidence of Use and Harms of Novel Benzodiazepines
ACMD Advisory Council on the Misuse of Drugs Novel Benzodiazepines A review of the evidence of use and harms of Novel Benzodiazepines April 2020 1 Contents 1. Introduction ................................................................................................................................. 4 2. Legal control of benzodiazepines .......................................................................................... 4 3. Benzodiazepine chemistry and pharmacology .................................................................. 6 4. Benzodiazepine misuse............................................................................................................ 7 Benzodiazepine use with opioids ................................................................................................... 9 Social harms of benzodiazepine use .......................................................................................... 10 Suicide ............................................................................................................................................. 11 5. Prevalence and harm summaries of Novel Benzodiazepines ...................................... 11 1. Flualprazolam ......................................................................................................................... 11 2. Norfludiazepam ....................................................................................................................... 13 3. Flunitrazolam .......................................................................................................................... -

Fatal Intoxication with U-47700 in Combination with Other NPS (N
Forensic Toxicology https://doi.org/10.1007/s11419-020-00568-1 CASE REPORT Fatal intoxication with U‑47700 in combination with other NPS (N‑ethylhexedrone, adinazolam, 4‑CIC, 4‑CMC) confrmed by identifcation and quantifcation in autopsy specimens and evidences Karolina Nowak1,2 · Paweł Szpot1,2 · Marcin Zawadzki1,2 Received: 12 September 2020 / Accepted: 22 December 2020 © The Author(s) 2021 Abstract Purpose We present a case of fatal intoxication with U-47700 in combination with other NPS (N-ethylhexedrone, adinazolam, 4-chloro-N-isopropylcathinone (4-CIC), 4-chloromethcathinone (4-CMC) and sertraline) confrmed by identifcation and quantifcation in biological materials and evidences found at the scene in 2017 in Poland. Methods Blood and urine samples were extracted with ethyl acetate from alkaline medium (pH 9); powders/crystals were diluted with methanol. The analysis was carried out using ultra-high-performance liquid chromatography–tandem mass spectrometry. Validation criteria were evaluated for blood and urine at the concentrations of 10 and 100 ng/mL. Results The validation parameters of the method were within acceptable ranges. In the presented case, the determined concentrations of drugs were as follows, in blood: U-47700, 1470 ng/mL; N-ethylhexedrone, 58 ng/mL; adinazolam, 18 ng/ mL; 4-CIC, 8.0 ng/mL; 4-CMC, 1.7 ng/mL; in urine: U-47700, 3940 ng/mL; N-ethylhexedrone, 147 ng/mL; adinazolam, 82 ng/mL; 4-CIC, 130 ng/mL; 4-CMC, 417 ng/mL. Sertraline (blood, 89 ng/mL; urine, 32 ng/mL) was also determined in both materials. The same substances were also found in 5 powders/crystals: U-47700 (12% by weight), N-ethylhexedrone (54%), adinazolam (14%), 4-CIC (23%), 4-CMC (26%). -
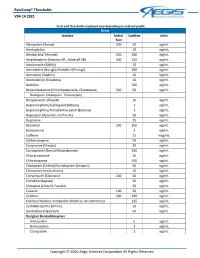
Paincomp® Thresholds V04.14.2021 Copyright © 2020 Aegis Sciences
PainComp® Thresholds V04.14.2021 Tests and thresholds employed vary depending on ordered profile Urine Analyte Initial Confirm Units Test Alprazolam (Xanax) 200 50 ng/mL Amitriptyline 10 ng/mL Amobarbital (Amytal) 200 200 ng/mL Amphetamine (Adzenys ER , Adderall XR) 500 250 ng/mL Aripiprazole (Abilify) 10 ng/mL Armodafinil (Nuvigil)/Modafinil (Provigil) 100 ng/mL Asenapine (Saphris) 10 ng/mL Atomoxetine (Strattera) 10 ng/mL Baclofen 500 ng/mL Benzodiazepines (Chlordiazepoxide, Clorazepate, 200 50 ng/mL Diazepam, Oxazepam, Temazepam) Brexpiprazole (Rexulti) 10 ng/mL Buprenorphine/Sublingual (Belbuca) 1 ng/mL Buprenorphine/Transdermal patch (Butrans) 1 ng/mL Bupropion (Aplenzin, Forfivo XL) 50 ng/mL Buspirone 25 ng/mL Butalbital 200 200 ng/mL Butorphanol 1 ng/mL Caffeine 15 mcg/mL Carbamazepine 50 ng/mL Cariprazine (Vraylar) 20 ng/mL Carisoprodol (Soma)/Meprobamate 200 ng/mL Chlorpromazine 10 ng/mL Chlorzoxazone 500 ng/mL Citalopram (Celexa)/Escitalopram (Lexapro) 50 ng/mL Clomipramine (Anafranil) 10 ng/mL Clonazepam (Klonopin) 200 50 ng/mL Clonidine (Kapvay) 50 ng/mL Clozapine (Clozaril, Fazaclo) 50 ng/mL Cocaine 100 50 ng/mL Codeine 200 100 ng/mL Cotinine/Nicotine metabolite (Habitrol, Nicoderm CQ) 125 ng/mL Cyclobenzaprine (Amrix) 10 ng/mL Dantrolene (Dantrium) 50 ng/mL Designer Benzodiazepines Adinazolam 5 ng/mL Bromazolam 1 ng/mL Clonazolam 1 ng/mL Copyright © 2020 Aegis Sciences Corporation All Rights Reserved PainComp® Thresholds V04.14.2021 Deschloroetizolam 1 ng/mL Diclazepam 1 ng/mL Etizolam 1 ng/mL Flualprazolam 1 ng/mL Flubromazepam -

Drug/Substance Trade Name(S)
A B C D E F G H I J K 1 Drug/Substance Trade Name(s) Drug Class Existing Penalty Class Special Notation T1:Doping/Endangerment Level T2: Mismanagement Level Comments Methylenedioxypyrovalerone is a stimulant of the cathinone class which acts as a 3,4-methylenedioxypyprovaleroneMDPV, “bath salts” norepinephrine-dopamine reuptake inhibitor. It was first developed in the 1960s by a team at 1 A Yes A A 2 Boehringer Ingelheim. No 3 Alfentanil Alfenta Narcotic used to control pain and keep patients asleep during surgery. 1 A Yes A No A Aminoxafen, Aminorex is a weight loss stimulant drug. It was withdrawn from the market after it was found Aminorex Aminoxaphen, Apiquel, to cause pulmonary hypertension. 1 A Yes A A 4 McN-742, Menocil No Amphetamine is a potent central nervous system stimulant that is used in the treatment of Amphetamine Speed, Upper 1 A Yes A A 5 attention deficit hyperactivity disorder, narcolepsy, and obesity. No Anileridine is a synthetic analgesic drug and is a member of the piperidine class of analgesic Anileridine Leritine 1 A Yes A A 6 agents developed by Merck & Co. in the 1950s. No Dopamine promoter used to treat loss of muscle movement control caused by Parkinson's Apomorphine Apokyn, Ixense 1 A Yes A A 7 disease. No Recreational drug with euphoriant and stimulant properties. The effects produced by BZP are comparable to those produced by amphetamine. It is often claimed that BZP was originally Benzylpiperazine BZP 1 A Yes A A synthesized as a potential antihelminthic (anti-parasitic) agent for use in farm animals. -

Benzodiazepine: Are They All the Same, Are They All Bad?
Benzodiazepine: Are they all the same, are they all bad? Jefferson B. Prince, MD Massachusetts General Hospital Harvard Medical School North Shore Medical Center [email protected] www.mghcme.org Disclosures If you have disclosures, state: “My spouse/partner and I have the following relevant financial relationship with a commercial interest to disclose: I am the author of the book “Almost Depressed” and have received payments from Harvard health Publications www.mghcme.org Benzodiazepines Augment the Effects of GABA • GABA is the main inhibitory neurotransmitter in the brain. • Benzodiazepines increase the affinity of GABA receptors for GABA; the effect of which increases Cl- conductance resulting in hyperpolarizing • Benzodiazepines exert their action only in the presence of GABA – for this reason they are called positive allosteric modulators (PAMs); acting as indirect agonists of the GABA receptor www.mghcme.org Benzodiazepines are Positive Allosteric Modulators (PAMs) of GABA-A Receptors • Allosteric sites are all receptor sites where GABA itself does not bind. • Allosteric modulators can be positive or negative. www.mghcme.org GABA Cells are Inter-neurons www.mghcme.org Selectivity for GABA-A Receptor Subunits GABA A Receptors containing Alpha 1 GABA A Receptors containing alpha 2 or subunits are involved in SLEEP MODULATION alpha 3 subunits are involved in EXPERIENCES of ANXIETY www.mghcme.org Existing Benzodiazepines are Non-selective • Benzodiazepines bind to GABA-A alpha subunits: 1, 2, 3 and alpha 5. • Each of subunits is associated with different effects • Benzodiazepines are/can be/cause: – Sedating – Anxiolytic – Muscle relaxation – Potentiate effects of Alcohol www.mghcme.org Benzodiazepines are Anxolytic & Hypnotic • ANXIOLYTIC EFFECT: By binding at GABA- A receptors in the amygdala, Benzodiazepines inhibit the activation of amygdala.