Thesis Reference
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Novel LRPPRC Compound Heterozygous Mutation in a Child
Piro et al. Italian Journal of Pediatrics (2020) 46:140 https://doi.org/10.1186/s13052-020-00903-7 CASE REPORT Open Access Novel LRPPRC compound heterozygous mutation in a child with early-onset Leigh syndrome French-Canadian type: case report of an Italian patient Ettore Piro1* , Gregorio Serra1, Vincenzo Antona1, Mario Giuffrè1, Elisa Giorgio2, Fabio Sirchia3, Ingrid Anne Mandy Schierz1, Alfredo Brusco2 and Giovanni Corsello1 Abstract Background: Mitochondrial diseases, also known as oxidative phosphorylation (OXPHOS) disorders, with a prevalence rate of 1:5000, are the most frequent inherited metabolic diseases. Leigh Syndrome French Canadian type (LSFC), is caused by mutations in the nuclear gene (2p16) leucine-rich pentatricopeptide repeat-containing (LRPPRC). It is an autosomal recessive neurogenetic OXPHOS disorder, phenotypically distinct from other types of Leigh syndrome, with a carrier frequency up to 1:23 and an incidence of 1:2063 in the Saguenay-Lac-St Jean region of Quebec. Recently, LSFC has also been reported outside the French-Canadian population. Patient presentation: We report a male Italian (Sicilian) child, born preterm at 28 + 6/7 weeks gestation, carrying a novel LRPPRC compound heterozygous mutation, with facial dysmorphisms, neonatal hypotonia, non-epileptic paroxysmal motor phenomena, and absent sucking-swallowing-breathing coordination requiring, at 4.5 months, a percutaneous endoscopic gastrostomy tube placement. At 5 months brain Magnetic Resonance Imagingshoweddiffusecortical atrophy, hypoplasia of corpus callosum, cerebellar vermis hypoplasia, and unfolded hippocampi. Both auditory and visual evoked potentials were pathological. In the following months Video EEG confirmed the persistence of sporadic non epileptic motor phenomena. No episode of metabolic decompensation, acidosis or ketosis, frequently observed in LSFC has been reported. -

The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma
cancers Review The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma Laura-Marie Ammer 1, Arabel Vollmann-Zwerenz 1, Viktoria Ruf 2, Christian H. Wetzel 3 , Markus J. Riemenschneider 4, Nathalie L. Albert 5, Philipp Beckhove 6 and Peter Hau 1,* 1 Wilhelm Sander-NeuroOncology Unit and Department of Neurology, University Hospital Regensburg, 93053 Regensburg, Germany; [email protected] (L.-M.A.); [email protected] (A.V.-Z.) 2 Center for Neuropathology and Prion Research, Ludwig Maximilians University of Munich, 81377 Munich, Germany; [email protected] 3 Molecular Neurosciences, Department of Psychiatry and Psychotherapy, University of Regensburg, 93053 Regensburg, Germany; [email protected] 4 Department of Neuropathology, Regensburg University Hospital, 93053 Regensburg, Germany; [email protected] 5 Department of Nuclear Medicine, Ludwig-Maximilians-University Munich, 81377 Munich, Germany; [email protected] 6 Regensburg Center for Interventional Immunology (RCI) and Department Internal Medicine III, University Hospital Regensburg, 93053 Regensburg, Germany; [email protected] * Correspondence: [email protected] Received: 14 September 2020; Accepted: 9 October 2020; Published: 14 October 2020 Simple Summary: The translocator protein (TSPO) has been under extensive investigation as a specific marker in positron emission tomography (PET) to visualize brain lesions following injury or disease. In recent years, TSPO is increasingly appreciated as a potential novel therapeutic target in cancer. In Glioblastoma (GBM), the most malignant primary brain tumor, TSPO expression levels are strongly elevated and scientific evidence accumulates, hinting at a pivotal role of TSPO in tumorigenesis and glioma progression. The aim of this review is to summarize the current literature on TSPO with respect to its role both in diagnostics and especially with regard to the critical hallmarks of cancer postulated by Hanahan and Weinberg. -

Building the Vertebrate Codex Using the Gene Breaking Protein Trap Library
Genetics, Development and Cell Biology Publications Genetics, Development and Cell Biology 2020 Building the vertebrate codex using the gene breaking protein trap library Noriko Ichino Mayo Clinic Gaurav K. Varshney National Institutes of Health Ying Wang Iowa State University Hsin-kai Liao Iowa State University Maura McGrail Iowa State University, [email protected] See next page for additional authors Follow this and additional works at: https://lib.dr.iastate.edu/gdcb_las_pubs Part of the Molecular Genetics Commons, and the Research Methods in Life Sciences Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ gdcb_las_pubs/261. For information on how to cite this item, please visit http://lib.dr.iastate.edu/howtocite.html. This Article is brought to you for free and open access by the Genetics, Development and Cell Biology at Iowa State University Digital Repository. It has been accepted for inclusion in Genetics, Development and Cell Biology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Building the vertebrate codex using the gene breaking protein trap library Abstract One key bottleneck in understanding the human genome is the relative under-characterization of 90% of protein coding regions. We report a collection of 1200 transgenic zebrafish strains made with the gene- break transposon (GBT) protein trap to simultaneously report and reversibly knockdown the tagged genes. Protein trap-associated mRFP expression shows previously undocumented expression of 35% and 90% of cloned genes at 2 and 4 days post-fertilization, respectively. Further, investigated alleles regularly show 99% gene-specific mRNA knockdown. -

(TSPO) in Mitochondrial Bioenergetics and Immune Processes
cells Review The Translocator Protein (TSPO) in Mitochondrial Bioenergetics and Immune Processes Calina Betlazar 1,2,* , Ryan J. Middleton 1 , Richard Banati 1,2 and Guo-Jun Liu 1,2,* 1 Human Health, Australian Nuclear Science and Technology Organisation, New Illawarra Road, Lucas Heights, NSW 2234, Australia; [email protected] (R.J.M.); [email protected] (R.B.) 2 Discipline of Medical Imaging & Radiation Sciences, Faculty of Medicine and Health, Brain and Mind Centre, University of Sydney, 94 Mallett Street, Camperdown, NSW 2050, Australia * Correspondence: [email protected] (C.B.); [email protected] (G-J.L.) Received: 11 January 2020; Accepted: 19 February 2020; Published: 24 February 2020 Abstract: The translocator protein (TSPO) is an outer mitochondrial membrane protein that is widely used as a biomarker of neuroinflammation, being markedly upregulated in activated microglia in a range of brain pathologies. Despite its extensive use as a target in molecular imaging studies, the exact cellular functions of this protein remain in question. The long-held view that TSPO plays a fundamental role in the translocation of cholesterol through the mitochondrial membranes, and thus, steroidogenesis, has been disputed by several groups with the advent of TSPO knockout mouse models. Instead, much evidence is emerging that TSPO plays a fundamental role in cellular bioenergetics and associated mitochondrial functions, also part of a greater role in the innate immune processes of microglia. In this review, we examine the more direct experimental literature surrounding the immunomodulatory effects of TSPO. We also review studies which highlight a more central role for TSPO in mitochondrial processes, from energy metabolism, to the propagation of inflammatory responses through reactive oxygen species (ROS) modulation. -
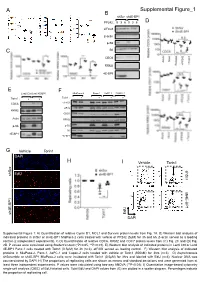
H Supplemental Figure 1 I D E B a C
A Supplemental Figure_1 B shScr sh4E-BP1 PP242 : 0 3 6 0 3 6 D eIF4G1 β-actin p-S6 C S6 CDC6 RRM2 4E-BP1 E F Lenti Ctrl Lenti 4E-BP1 MiaPaca-2 Panc-1 AsPC-1 Capan-2 Torin1 : Torin1 : + + + + + + CDC6 eIF4G1 eIF3a RRM2 CDC6 Actin RRM2 p-S6 p-S6 S6 4E-BP1 4E-BP1 G Vehicle Torin1 DAPI H I Vehicle Torin1 62.20% 45.30% shScr sh4E-BP1 EdU shScr 59.58% 49.47% EdU sh4E-BP1 DAPI Supplemental Figure 1: A) Quantification of relative Cyclin D1, MCL1 and Survivin protein levels from Fig. 1A. B) Western blot analysis of indicated proteins in shScr or sh4E-BP1 MiaPaca-2 cells treated with vehicle or PP242 (5µM) for 3h and 6h. β-actin served as a loading control (2 independent experiments). C-D) Quantification of relative CDC6, RRM2 and CDC7 protein levels from (C) Fig. 2C and (D) Fig. 2D. P values were calculated using Student’s t-test (*P<0.05, **P<0.01). E) Western blot analysis of indicated proteins in Lenti Ctrl or Lenti 4E-BP1 Panc-1 cells treated with Torin1 (0.5µM) for 3h (n=3). eIF4GI served as loading control. F) Western blot analysis of indicated proteins in MiaPaca-2, Panc-1, AsPC-1 and Capan-2 cells treated with vehicle or Torin1 (500nM) for 3hrs (n=3). G) Asynchronous shScramble or sh4E-BP1 MiaPaca-2 cells were incubated with Torin1 (0.5μM) for 3hrs and labeled with EdU (n=3). Nuclear DNA was counterstained by DAPI. H) The proportions of replicating cells are shown as means and standard deviations and were generated from at least three independent experiments. -

De Novo Neurosteroidogenesis in Human Microglia: Involvement of the 18 Kda Translocator Protein
International Journal of Molecular Sciences Article De novo Neurosteroidogenesis in Human Microglia: Involvement of the 18 kDa Translocator Protein Lorenzo Germelli 1,† , Eleonora Da Pozzo 1,† , Chiara Giacomelli 1 , Chiara Tremolanti 1 , Laura Marchetti 1, Christian H. Wetzel 2 , Elisabetta Barresi 1 , Sabrina Taliani 1 , Federico Da Settimo 1 , Claudia Martini 1,* and Barbara Costa 1 1 Department of Pharmacy, University of Pisa, 56126 Pisa, Italy; [email protected] (L.G.); [email protected] (E.D.P.); [email protected] (C.G.); [email protected] (C.T.); [email protected] (L.M.); [email protected] (E.B.); [email protected] (S.T.); [email protected] (F.D.S.); [email protected] (B.C.) 2 Department of Psychiatry and Psychotherapy, Molecular Neurosciences, University of Regensburg, 93053 Regensburg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +3905-0221-9523 † These authors contributed equally to this work. Abstract: Neuroactive steroids are potent modulators of microglial functions and are capable of counteracting their excessive reactivity. This action has mainly been ascribed to neuroactive steroids released from other sources, as microglia have been defined unable to produce neurosteroids de novo. Unexpectedly, immortalized murine microglia recently exhibited this de novo biosynthesis; herein, de novo neurosteroidogenesis was characterized in immortalized human microglia. The results demonstrated that C20 and HMC3 microglial cells constitutively express members of the Citation: Germelli, L.; Da Pozzo, E.; neurosteroidogenesis multiprotein machinery—in particular, the transduceosome members StAR Giacomelli, C.; Tremolanti, C.; and TSPO, and the enzyme CYP11A1. Moreover, both cell lines produce pregnenolone and transcrip- Marchetti, L.; Wetzel, C.H.; Barresi, E.; tionally express the enzymes involved in neurosteroidogenesis. -

Tu Cornellgrad 0058F 10421.Pdf (5.371Mb)
MOLECULAR FUNCTION OF MAMMALIAN TRANSLOCATOR PROTEIN (TSPO) A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Lan Ngoc Ly Tu August 2017 © 2017 Lan Ngoc Ly Tu MOLECULAR FUNCTION OF MAMMALIAN TRANSLOCATOR PROTEIN (TSPO) Lan Ngoc Ly Tu, Ph. D. Cornell University 2017 Translocator protein (TSPO), previously known as the peripheral benzodiazepine receptor (PBR), is a mitochondrial outer membrane protein highly conserved from bacteria to humans. TSPO is found to be upregulated in many pathological conditions, making it an attractive target for both diagnostic and therapeutic purposes in human medicine. However, the exact molecular function of TSPO remained unclear. For the past 25 years, TSPO was depicted to transport cholesterol from the cytosol to the inner mitochondrial membrane, the rate-limiting step in steroid hormone biosynthesis. Its critical role in survival and development was reinforced by a report that claimed TSPO knockout (Tspo-/-) mice were embryonic lethal. Therefore, there had been no genetic models to study the function of TSPO and all functional interpretations of TSPO were based on in vitro experiments using pharmacological tools. Our lab generated the first global Tspo-/- mice which surprisingly were healthy with no apparent abnormalities. Deletion of TSPO in different steroidogenic cell lines also did not affect cell viability. Furthermore, steroid hormone production was not affected in Tspo-/- mice or in steroidogenic cells lacking TSPO compared to the controls, indicating that TSPO is not involved in steroidogenesis. The stimulating effect of some TSPO binding chemicals on steroid hormone production that formed the early basis for linking TSPO and steroidogenesis was also found to be inaccurate. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Characterization and Proteomic-Transcriptomic Investigation of Monocarboxylate Transporter 6 Knockout Mice
Supplemental material to this article can be found at: http://molpharm.aspetjournals.org/content/suppl/2019/07/10/mol.119.116731.DC1 1521-0111/96/3/364–376$35.00 https://doi.org/10.1124/mol.119.116731 MOLECULAR PHARMACOLOGY Mol Pharmacol 96:364–376, September 2019 Copyright ª 2019 by The American Society for Pharmacology and Experimental Therapeutics Characterization and Proteomic-Transcriptomic Investigation of Monocarboxylate Transporter 6 Knockout Mice: Evidence of a Potential Role in Glucose and Lipid Metabolism s Robert S. Jones, Chengjian Tu, Ming Zhang, Jun Qu, and Marilyn E. Morris Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, University at Buffalo, State University of New York, Buffalo, New York (R.S.J., C.T., J.Q., M.E.M.); and New York State Center of Excellence in Bioinformatics and Life Sciences, Buffalo, New York (C.T., M.Z., J.Q.) Received March 21, 2019; accepted June 27, 2019 Downloaded from ABSTRACT Monocarboxylate transporter 6 [(MCT6), SLC16A5] is an or- Mct62/2 mice demonstrated significant changes in 199 genes phan transporter with no known endogenous substrates or in the liver compared with Mct61/1 mice. In silico biological physiological role. Previous in vitro and in vivo experiments pathway analyses revealed significant changes in proteins and molpharm.aspetjournals.org investigated MCT6 substrate/inhibitor specificity in Xenopus genes involved in glucose and lipid metabolism–associated laevis oocytes; however, these data remain limited. Transcrip- pathways. This study is the first to provide evidence for an tomic changes in the livers of mice undergoing different dieting association of Mct6 in the regulation of glucose and lipid schemes have suggested that Mct6 plays a role in glucose metabolism. -

Gene Ontology Functional Annotations and Pleiotropy
Network based analysis of genetic disease associations Sarah Gilman Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2013 Sarah Gilman All Rights Reserved ABSTRACT Network based analysis of genetic disease associations Sarah Gilman Despite extensive efforts and many promising early findings, genome-wide association studies have explained only a small fraction of the genetic factors contributing to common human diseases. There are many theories about where this “missing heritability” might lie, but increasingly the prevailing view is that common variants, the target of GWAS, are not solely responsible for susceptibility to common diseases and a substantial portion of human disease risk will be found among rare variants. Relatively new, such variants have not been subject to purifying selection, and therefore may be particularly pertinent for neuropsychiatric disorders and other diseases with greatly reduced fecundity. Recently, several researchers have made great progress towards uncovering the genetics behind autism and schizophrenia. By sequencing families, they have found hundreds of de novo variants occurring only in affected individuals, both large structural copy number variants and single nucleotide variants. Despite studying large cohorts there has been little recurrence among the genes implicated suggesting that many hundreds of genes may underlie these complex phenotypes. The question -

(Cos) Rnas and of a Conserved Family of Organellar RNA-Binding Proteins, the Heptatricopeptide Repeat Proteins, in the Malaria Parasite Arne Hillebrand1, Joachim M
Published online 8 August 2018 Nucleic Acids Research, 2018, Vol. 46, No. 19 10417–10431 doi: 10.1093/nar/gky710 Identification of clustered organellar short (cos) RNAs and of a conserved family of organellar RNA-binding proteins, the heptatricopeptide repeat proteins, in the malaria parasite Arne Hillebrand1, Joachim M. Matz2, Martin Almendinger1,KatjaMuller¨ 2, Kai Matuschewski2 and Christian Schmitz-Linneweber1,* 1Humboldt University Berlin, Molecular Genetics, Berlin, Germany and 2Humboldt University, Department of Molecular Parasitology, Berlin, Germany Received December 12, 2017; Revised July 20, 2018; Editorial Decision July 23, 2018; Accepted July 24, 2018 ABSTRACT malaria globally, resulting in almost half a million deaths (1). While some antimalarial treatments are available, para- Gene expression in mitochondria of Plasmodium site resistance is a continuing challenge. Plasmodium spp. falciparum is essential for parasite survival. The belong to the family of apicomplexan parasites, most of molecular mechanisms of Plasmodium organellar which contain a non-photosynthetic plastid called the api- gene expression remain poorly understood. This coplast (2). As a remnant of a red algae chloroplast, the api- includes the enigmatic assembly of the mitochon- coplast contains its own DNA, as does the Plasmodium mi- drial ribosome from highly fragmented rRNAs. Here, tochondrion. The Plasmodium nuclear genome has a size we present the identification of clustered organellar of 24 MB and contains >5000 genes; by contrast, the api- short RNA fragments (cosRNAs) that are possible coplast and mitochondrial genomes are small––35 and 6 kb, footprints of RNA-binding proteins (RBPs) in Plas- respectively. modium organelles. In plants, RBPs of the pentatri- In keeping with the descendance of the apicoplast from cyanobacteria, the apicoplast genome is organized similarly copeptide repeat (PPR) class produce footprints as a to bacterial chromosomes.