BARBITURATES, PHENYTOIN and CARBAMAZEPINE by SOLID PHASE EXTRACTION and HIGH PERFORMANCE LIQUID CHROMATOGRAPHY
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metabolic-Hydroxy and Carboxy Functionalization of Alkyl Moieties in Drug Molecules: Prediction of Structure Influence and Pharmacologic Activity
molecules Review Metabolic-Hydroxy and Carboxy Functionalization of Alkyl Moieties in Drug Molecules: Prediction of Structure Influence and Pharmacologic Activity Babiker M. El-Haj 1,* and Samrein B.M. Ahmed 2 1 Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, University of Science and Technology of Fujairah, Fufairah 00971, UAE 2 College of Medicine, Sharjah Institute for Medical Research, University of Sharjah, Sharjah 00971, UAE; [email protected] * Correspondence: [email protected] Received: 6 February 2020; Accepted: 7 April 2020; Published: 22 April 2020 Abstract: Alkyl moieties—open chain or cyclic, linear, or branched—are common in drug molecules. The hydrophobicity of alkyl moieties in drug molecules is modified by metabolic hydroxy functionalization via free-radical intermediates to give primary, secondary, or tertiary alcohols depending on the class of the substrate carbon. The hydroxymethyl groups resulting from the functionalization of methyl groups are mostly oxidized further to carboxyl groups to give carboxy metabolites. As observed from the surveyed cases in this review, hydroxy functionalization leads to loss, attenuation, or retention of pharmacologic activity with respect to the parent drug. On the other hand, carboxy functionalization leads to a loss of activity with the exception of only a few cases in which activity is retained. The exceptions are those groups in which the carboxy functionalization occurs at a position distant from a well-defined primary pharmacophore. Some hydroxy metabolites, which are equiactive with their parent drugs, have been developed into ester prodrugs while carboxy metabolites, which are equiactive to their parent drugs, have been developed into drugs as per se. -

Chronic Use of Opioid Medications Before and After Bariatric Surgery
Supplementary Online Content Raebel MA, Newcomer SR, Reifler LM, et al. Chronic use of opioid medications before and after bariatric surgery. JAMA. doi:10.1001/jama.2013.278344 eFigure. Opioid Dispensings in 60 Days Before and 60 Days After Bariatric Surgery eTable 1. Opioid Classification and Morphine Equivalents Conversion Factors for Opioids Administered by Tablet, Capsule, Liquid, Transdermal Patch, and Transmucosal Formulations eTable 2. ICD-9 Codes for Comorbid Diagnosis of Substance Abuse, Anxiety, Posttraumatic Stress Disorder, and/or Depression eTable 3. ICD-9 Codes for Inclusion and Exclusion of Chronic Pain Diagnoses eTable 4. Therapeutic Classes, Generic Names, and Selected Brand Names of Covariate Medications eTable 5. Types of Opioids Dispensed During the Year Before and the Year After Bariatric Surgery Among Presurgery Chronic Opioid Users eTable 6. Types of Opioids Used Before and After Bariatric Surgery Among 933 Individuals With Chronic Opioid Use Before Bariatric Surgery eTable 7. Unadjusted Characteristics of Chronic Opioid Users According to Whether or Not Both Presurgery and Postsurgery Body Mass Indexes (BMIs) Data Were Available eTable 8. Presurgery and Postsurgery Use of Opioids and Selected Other Analgesic and Adjunctive Pain Medication Classes Among Presurgery Chronic Opioid Users With Presurgery Chronic Pain Diagnoses This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: -

BUTALBITAL ANALGESICS Allzital (Butalbital-Acetaminophen
BUTALBITAL ANALGESICS Allzital (butalbital-acetaminophen), butalbital-aspirin-caffeine, butalbital-aspirin-caffeine- codeine, butalbital-acetaminophen, butalbital-acetaminophen-caffeine, butalbital- acetaminophen-caffeine-codeine, Vanatol LQ (butalbital-acetaminophen-caffeine liquid oral solution) RATIONALE FOR INCLUSION IN PA PROGRAM Background Butalbital containing products are non-opioid analgesics that contain a combination of different drug products indicated for the relief of the symptom complex of tension (or muscle contraction) headache pain. Butalbital is a short to intermediate-acting barbiturate that works in concert with acetaminophen, an antipyretic non-salicylate agent, aspirin, a pain-relieving NSAID, and caffeine, a stimulant that works in the CNS, to decrease pain via a mechanism that isn’t well understood. Butalbital is a habit-forming drug that potentiates the effects of other commonly abuse drugs or substances like alcohol. Caffeine might help increase vasodilation and smooth muscle relaxation, while butalbital is thought to help balance the CNS stimulation caused by caffeine and produces depressant effects (1). Regulatory Status FDA approved indication: Butalbital containing products are used in the relief of the symptom complex of tension or muscle contraction headaches (2-8). Frequent use of acute medications is generally thought to cause medication-overuse headache. To decrease the risk of medication-overuse headache (“rebound headache” or “drug-induced headache”) many experts limit acute therapy to two headache days per week on a regular basis. Based on concerns of overuse, medication-overuse headache, and withdrawal, the use of butalbital-containing analgesics should be limited and carefully monitored. The Allzital limit is set to the maximum of 12 doses per day for acute treatment of 8 headaches per month as this product contains less butalbital than other products. -

And Calcium, Magnesium, Potassium and Sodium Oxybates (Xywav™)
Louisiana Medicaid Sodium Oxybate (Xyrem®) and Calcium, Magnesium, Potassium and Sodium Oxybates (Xywav™) The Louisiana Uniform Prescription Drug Prior Authorization Form should be utilized to request clinical authorization for sodium oxybate (Xyrem®) and calcium, magnesium, potassium and sodium oxybates (Xywav™). These agents have Black Box Warnings and are subject to Risk Evaluation and Mitigation Strategy (REMS) under FDA safety regulations. Please refer to individual prescribing information for details. Approval Criteria • The recipient is 7 years of age or older on date of request; AND • The recipient has a documented diagnosis of narcolepsy or cataplexy; AND • The prescribing provider is a Board-Certified Neurologist or a Board-Certified Sleep Medicine Physician; AND • By submitting the authorization request, the prescriber attests to the following: o The prescribing information for the requested medication has been thoroughly reviewed, including any Black Box Warning, Risk Evaluation and Mitigation Strategy (REMS), contraindications, minimum age requirements, recommended dosing, and prior treatment requirements; AND o All laboratory testing and clinical monitoring recommended in the product prescribing information have been completed as of the date of the request and will be repeated as recommended; AND o The recipient has no inappropriate concomitant drug therapies or disease states. Duration of initial authorization approval: 3 months Reauthorization Criteria • The recipient continues to meet all initial approval criteria; AND -

Prescription Drug Management
Check out our new site: www.acllaboratories.com Prescription Drug Management Non Adherence, Drug Misuse, Increased Healthcare Costs Reports from the Centers for DiseasePrescription Control and Prevention (CDC) say Drug deaths from Managementmedication overdose have risen for 11 straight years. In 2008 more than 36,000 people died from drug overdoses, and most of these deaths were caused by prescription Nondrugs. Adherence,1 Drug Misuse, Increased Healthcare Costs The CDC analysis found that nearly 40,000 drug overdose deaths were reported in 2010. Prescribed medication accounted for almost 60 percent of the fatalities—far more than deaths from illegal street drugs. Abuse of painkillers like ReportsOxyContin from and the VicodinCenters forwere Disease linked Control to the and majority Prevention of the (CDC) deaths, say deaths from according to the report.1 medication overdose have risen for 11 straight years. In 2008 more than 36,000 people died from drug overdoses, and most of these deaths were caused by prescription drugs. 1 A health economics study analyzed managed care claims of more than 18 million patients, finding that patients undergoing opioid therapyThe CDCfor chronic analysis pain found who that may nearly not 40,000 be following drug overdose their prescription deaths were regimenreported in 2010. Prescribed medication accounted for almost 60 percent of the fatalities—far more than deaths have significantly higher overall healthcare costs. from illegal street drugs. Abuse of painkillers like OxyContin and Vicodin were linked to the majority of the deaths, according to the report.1 ACL offers drug management testing to provide information that can aid clinicians in therapy and monitoring to help improve patientA health outcomes. -

Additional Requested Drugs
PART 1 PART PATIENT PRESCRIBED MEDICATIONS: o ACTIQ o DESIPRAMINE o HYDROCODONE o MORPHINE o ROXICODONE o ADDERALL o DIAZEPAM* o HYDROMORPHONE o MS CONTIN o SOMA o ALPRAZOLAM* o DILAUDID o IMIPRAMINE o NEURONTIN o SUBOXONE o AMBIEN o DURAGESIC o KADIAN o NORCO o TEMAZEPAM o AMITRIPTYLINE o ELAVIL o KETAMINE o NORTRIPTYLINE o TRAMADOL* o ATIVAN o EMBEDA o KLONOPIN o NUCYNTA o TYLENOL #3 o AVINZA o ENDOCET o LORAZEPAM o OPANA o ULTRAM o BUPRENEX o FENTANYL* o LORTAB o OXYCODONE o VALIUM o BUPRENORPHINE o FIORICET o LORCET o OXYCONTIN o VICODIN o BUTRANS o GABAPENTIN o LYRICA o PERCOCET o XANAX o CLONAZEPAM* o GRALISE o METHADONE o RESTORIL TO RE-ORDER CALL RITE-PRINT 718/384-4288 RITE-PRINT CALL TO RE-ORDER ALLIANCE DRUG PANEL (SEE BELOW) o ALCOHOL o BARBITURATES o ILLICITS o MUSCLE RELAXANTS o OPIODS (SYN) ETHANOL PHENOBARBITAL 6-MAM (HEROIN)* CARISPRODOL FENTANYL* BUTABARBITAL a-PVP MEPROBAMATE MEPERIDINE o AMPHETAMINES SECOBARBITAL BENZOYLECGONINE NALOXONE AMPHETAMINE PENTOBARBITAL LSD o OPIODS (NATURAL) METHADONE* METHAMPHETAMINE BUTALBITAL MDA CODEINE METHYLPHENEDATE MDEA MORPHINE o NON-OPIOID o BENZODIAZEPINES MDMA ANALGESICS o OPIODS (SEMI-SYN) o ANTICONVULSIVES ALPRAZOLAM* MDPV TRAMADOL BUPRENORPHINE* GABAPENTIN CLONAZEPAM* MEPHEDRONE TAPENTADOL DIHROCODEINE PREGABALIN DIAZEPAM METHCATHINONE DESOMORPHINE o NON-BENZODIAZEPINE FLUNITRAZEPAM* METHYLONE HYDROCODONE HYPNOTIC o ANTIDEPRESSANTS FLURAZEPAM* PCP HYDROMORPHONE ZOLPIDEM* AMITRIPTYLINE LORAZEPAM THC* OXYCODONE DOXEPIN OXAZEPAM CBD ° OXYMORPHONE o MISCELLANEOUS DRUGS IMIPRIMINE -

Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules
Medication Guide Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules, C III Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules are: • A strong prescription pain medicine that contains an opioid (narcotic) that is indicated for the relief of the symptom complex of tension (or muscle contraction) headache, when other pain treatments such as non-opioid pain medicines do not treat your pain well enough or you cannot tolerate them. • An opioid pain medicine that can put you at risk for overdose and death. Even if you take your dose correctly as prescribed you are at risk for opioid addiction, abuse, and misuse that can lead to death. Important information about Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules: • Get emergency help right away if you take too much Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules (overdose). When you first start taking Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules, when your dose is changed, or if you take too much (overdose), serious or life-threatening breathing problems that can lead to death may occur. • Taking Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules with other opioid medicines, benzodiazepines, alcohol, or other central nervous system depressants (including street drugs) can cause severe drowsiness, decreased awareness, breathing problems, coma, and death. • Never give anyone else your Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules. They could die from taking it. Store Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules away from children and in a safe place to prevent stealing or abuse. Selling or giving away Butalbital, Acetaminophen, Caffeine, and Codeine Phosphate Capsules is against the law. • Get emergency help right away if you take more than 4,000 mg of acetaminophen in 1 day. -
![Nembutal [Pentobarbital] Injection and Seconal [Secobarbital] Capsules](https://docslib.b-cdn.net/cover/6349/nembutal-pentobarbital-injection-and-seconal-secobarbital-capsules-686349.webp)
Nembutal [Pentobarbital] Injection and Seconal [Secobarbital] Capsules
Pharmacy Benefit Coverage Criteria Effective Date .......................................... 12/1/2020 Next Review Date… ................................... 12/1/2021 Coverage Policy Number ................................ P0095 Nembutal [pentobarbital] injection and Seconal [secobarbital] capsules Table of Contents Related Coverage Resources Overview .............................................................. 1 Coverage Policy ................................................... 1 FDA Summary ..................................................... 1 General Background ............................................ 2 References .......................................................... 2 INSTRUCTIONS FOR USE The following Coverage Policy applies to health benefit plans administered by Cigna Companies. Certain Cigna Companies and/or lines of business only provide utilization review services to clients and do not make coverage determinations. References to standard benefit plan language and coverage determinations do not apply to those clients. Coverage Policies are intended to provide guidance in interpreting certain standard benefit plans administered by Cigna Companies. Please note, the terms of a customer’s particular benefit plan document [Group Service Agreement, Evidence of Coverage, Certificate of Coverage, Summary Plan Description (SPD) or similar plan document] may differ significantly from the standard benefit plans upon which these Coverage Policies are based. For example, a customer’s benefit plan document may contain a specific -
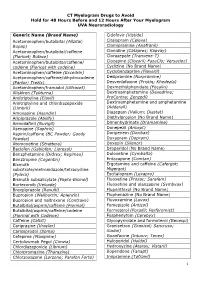
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology Generic Name (Brand Name) Cidofovir (Vistide) Acetaminophen/butalbital (Allzital; Citalopram (Celexa) Bupap) Clomipramine (Anafranil) Acetaminophen/butalbital/caffeine Clonidine (Catapres; Kapvay) (Fioricet; Butace) Clorazepate (Tranxene-T) Acetaminophen/butalbital/caffeine/ Clozapine (Clozaril; FazaClo; Versacloz) codeine (Fioricet with codeine) Cyclizine (No Brand Name) Acetaminophen/caffeine (Excedrin) Cyclobenzaprine (Flexeril) Acetaminophen/caffeine/dihydrocodeine Desipramine (Norpramine) (Panlor; Trezix) Desvenlafaxine (Pristiq; Khedezla) Acetaminophen/tramadol (Ultracet) Dexmethylphenidate (Focalin) Aliskiren (Tekturna) Dextroamphetamine (Dexedrine; Amitriptyline (Elavil) ProCentra; Zenzedi) Amitriptyline and chlordiazepoxide Dextroamphetamine and amphetamine (Limbril) (Adderall) Amoxapine (Asendin) Diazepam (Valium; Diastat) Aripiprazole (Abilify) Diethylpropion (No Brand Name) Armodafinil (Nuvigil) Dimenhydrinate (Dramamine) Asenapine (Saphris) Donepezil (Aricept) Aspirin/caffeine (BC Powder; Goody Doripenem (Doribax) Powder) Doxapram (Dopram) Atomoxetine (Strattera) Doxepin (Silenor) Baclofen (Gablofen; Lioresal) Droperidol (No Brand Name) Benzphetamine (Didrex; Regimex) Duloxetine (Cymbalta) Benztropine (Cogentin) Entacapone (Comtan) Bismuth Ergotamine and caffeine (Cafergot; subcitrate/metronidazole/tetracycline Migergot) (Pylera) Escitalopram (Lexapro) Bismuth subsalicylate (Pepto-Bismol) Fluoxetine (Prozac; Sarafem) -

HS 172 R5/13 Briefly Review the Objectives, Content and Activities of This Session
HS 172 R5/13 Briefly review the objectives, content and activities of this session. Upon successfully completing this session the participant will be able to: • Explain a brief history of the CNS Depressant category of drugs. • Identify common drug names and terms associated with this category. • Identify common methods of administration for this category. • Describe the symptoms, observable signs and other effects associated with this category. CONTENT SEGMENTS LEARNING ACTIVITIES A. Overview of the Category Instructor-Led Presentations B. Possible Effects Instructor Led Demonstrations C. OtdDtifEfftOnset and Duration of Effects RdiAiReading Assignmen ts D. Overdose Signs and Symptoms Video Presentations E. Expected Results of the Evaluation Slide Presentations F. Classification Exemplar HS 172 R5/13 9-2 • Explain the typical time parameters, i.e. onset and duration of effects, associated with this category. • List the clues that are likely to emerge when the drug influence evaluation is conducted for a person under the influence of this category of drugs. • Correctly answer the “topics for study” questions at the end of this session. HS 172 R5/13 9-3 A. Overview of the Category CNS Depressants Central Nervous System Depressants slow down the operations of the brain. Point out that other common names for CNS Depressants are “downers” and “sedative-hypnotics.” • Depressants first affect those arareaseas of the brain that control a person’ s conscious, voluntary actions. • Judgment, inhibitions and reaction time are some of the things that CNS Depressants affect first. • As the dose is increased, depressants begin to affect the parts of the brain that control the body’s automatic processes, heartbeat, respiration, etc. -

2019 01 07 Classification Substances
DRUG TESTING STANDARDS AND PRACTICES PROGRAM. Uniform Classification Guidelines for Foreign Substances And Recommended Penalties Model Rule. January, 2019 (V.14.0) © ASSOCIATION OF RACING COMMISSIONERS INTERNATIONAL – 2019. Association of Racing Commissioners International 2365 Harrodsburg Road- B450 Lexington, Kentucky, USA www.arci.com Page 1 of 66 Preamble to the Uniform Classification Guidelines of Foreign Substances The Preamble to the Uniform Classification Guidelines was approved by the RCI Drug Testing and Quality Assurance Program Committee (now the Drug Testing Standards and Practices Program Committee) on August 26, 1991. Minor revisions to the Preamble were made by the Drug Classification subcommittee (now the Veterinary Pharmacologists Subcommittee) on September 3, 1991. "The Uniform Classification Guidelines printed on the following pages are intended to assist stewards, hearing officers and racing commissioners in evaluating the seriousness of alleged violations of medication and prohibited substance rules in racing jurisdictions. Practicing equine veterinarians, state veterinarians, and equine pharmacologists are available and should be consulted to explain the pharmacological effects of the drugs listed in each class prior to any decisions with respect to penalities to be imposed. The ranking of drugs is based on their pharmacology, their ability to influence the outcome of a race, whether or not they have legitimate therapeutic uses in the racing horse, or other evidence that they may be used improperly. These classes of drugs are intended only as guidelines and should be employed only to assist persons adjudicating facts and opinions in understanding the seriousness of the alleged offenses. The facts of each case are always different and there may be mitigating circumstances which should always be considered. -

First Reported Case of Lorazepam-Assisted Interview in A
Hindawi Publishing Corporation Case Reports in Psychiatry Volume 2014, Article ID 346939, 4 pages http://dx.doi.org/10.1155/2014/346939 Case Report First Reported Case of Lorazepam-Assisted Interview in a Young Indian Female Presenting with Dissociative Identity Disorder and Improvement in Symptoms after the Interview Raheel Mushtaq,1,2 Sheikh Shoib,1,2 Tasleem Arif,3 Tabindah Shah,4 and Sahil Mushtaq5 1 ECT Clinic, Postgraduate Department of Psychiatry, Government Medical College, Srinagar, Jammu and Kashmir 190010, India 2 Memory Clinic, Postgraduate Department of Psychiatry, Government Medical College, Srinagar 190010, India 3 Post Graduate Department of Dermatology, Government Medical College, Srinagar 190010, India 4 Government Medical College, Srinagar, Jammu and Kashmir 190010, India 5 Acharya Shri Chander College of Medical Sciences, Jammu, India Correspondence should be addressed to Raheel Mushtaq; [email protected] Received 30 May 2014; Revised 17 July 2014; Accepted 18 July 2014; Published 5 August 2014 Academic Editor: Toshiya Inada Copyright © 2014 Raheel Mushtaq et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Dissociative identity disorder (DID) is one of the most fascinating disorders in psychiatry. The arduous search to reveal the obscurity of this disorder has led to colossal research in this area over the years. Although drug-assisted interviews are not widely used, they may be beneficial for some patients that do not respond to conventional treatments such as supportive psychotherapy or psychopharmacotherapy. Drug-assisted interviews facilitate recall of memories in promoting integration of dissociative information.