Adulthood Asthma As a Consequence of Childhood Adversity: a Systematic Review of Epigenetically Affected Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Integrative Genomic and Epigenomic Analyses Identified IRAK1 As a Novel Target for Chronic Inflammation-Driven Prostate Tumorigenesis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.16.447920; this version posted June 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Integrative genomic and epigenomic analyses identified IRAK1 as a novel target for chronic inflammation-driven prostate tumorigenesis Saheed Oluwasina Oseni1,*, Olayinka Adebayo2, Adeyinka Adebayo3, Alexander Kwakye4, Mirjana Pavlovic5, Waseem Asghar5, James Hartmann1, Gregg B. Fields6, and James Kumi-Diaka1 Affiliations 1 Department of Biological Sciences, Florida Atlantic University, Florida, USA 2 Morehouse School of Medicine, Atlanta, Georgia, USA 3 Georgia Institute of Technology, Atlanta, Georgia, USA 4 College of Medicine, Florida Atlantic University, Florida, USA 5 Department of Computer and Electrical Engineering, Florida Atlantic University, Florida, USA 6 Department of Chemistry & Biochemistry and I-HEALTH, Florida Atlantic University, Florida, USA Corresponding Author: [email protected] (S.O.O) Running Title: Chronic inflammation signaling in prostate tumorigenesis bioRxiv preprint doi: https://doi.org/10.1101/2021.06.16.447920; this version posted June 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract The impacts of many inflammatory genes in prostate tumorigenesis remain understudied despite the increasing evidence that associates chronic inflammation with prostate cancer (PCa) initiation, progression, and therapy resistance. -

Expression Gene Network Analyses Reveal Molecular Mechanisms And
www.nature.com/scientificreports OPEN Diferential expression and co- expression gene network analyses reveal molecular mechanisms and candidate biomarkers involved in breast muscle myopathies in chicken Eva Pampouille1,2, Christelle Hennequet-Antier1, Christophe Praud1, Amélie Juanchich1, Aurélien Brionne1, Estelle Godet1, Thierry Bordeau1, Fréderic Fagnoul2, Elisabeth Le Bihan-Duval1 & Cécile Berri1* The broiler industry is facing an increasing prevalence of breast myopathies, such as white striping (WS) and wooden breast (WB), and the precise aetiology of these occurrences remains poorly understood. To progress our understanding of the structural changes and molecular pathways involved in these myopathies, a transcriptomic analysis was performed using an 8 × 60 K Agilent chicken microarray and histological study. The study used pectoralis major muscles from three groups: slow-growing animals (n = 8), fast-growing animals visually free from defects (n = 8), or severely afected by both WS and WB (n = 8). In addition, a weighted correlation network analysis was performed to investigate the relationship between modules of co-expressed genes and histological traits. Functional analysis suggested that selection for fast growing and breast meat yield has progressively led to conditions favouring metabolic shifts towards alternative catabolic pathways to produce energy, leading to an adaptive response to oxidative stress and the frst signs of infammatory, regeneration and fbrosis processes. All these processes are intensifed in muscles afected by severe myopathies, in which new mechanisms related to cellular defences and remodelling seem also activated. Furthermore, our study opens new perspectives for myopathy diagnosis by highlighting fne histological phenotypes and genes whose expression was strongly correlated with defects. Te poultry industry relies on the production of fast-growing chickens, which are slaughtered at high weights and intended for cutting and processing. -

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

Product Data Sheet
Product Data Sheet ExProfileTM Human AMPK Signaling Related Gene qPCR Array For focused group profiling of human AMPK signaling genes expression Cat. No. QG004-A (4 x 96-well plate, Format A) Cat. No. QG004-B (4 x 96-well plate, Format B) Cat. No. QG004-C (4 x 96-well plate, Format C) Cat. No. QG004-D (4 x 96-well plate, Format D) Cat. No. QG004-E (4 x 96-well plate, Format E) Plates available individually or as a set of 6. Each set contains 336 unique gene primer pairs deposited in one 96-well plate. Introduction The ExProfile human AMPK signaling related gene qPCR array profiles the expression of 336 human genes related to AMPK-mediated signal transduction. These genes are carefully chosen for their close pathway correlation based on a thorough literature search of peer-reviewed publications, mainly including genes that encode AMP-activated protein kinase complex,its regulators and targets involved in many important biological processes, such as glucose uptake, β-oxidation of fatty acids and modulation of insulin secretion. This array allows researchers to study the pathway-related genes to gain understanding of their roles in the different biological processes. QG004 plate 01: 84 unique gene PCR primer pairs QG004 plate 02: 84 unique gene PCR primer pairs QG004 plate 03: 84 unique gene PCR primer pairs QG004 plate 04: 84 unique gene PCR primer pairs Shipping and storage condition Shipped at room temperate Stable for at least 6 months when stored at -20°C Array format GeneCopoeia provides five qPCR array formats (A, B, C, D, and E) suitable for use with the following real- time cyclers. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Disrupted Iron Homeostasis Causes Dopaminergic Neurodegeneration in Mice
Disrupted iron homeostasis causes dopaminergic neurodegeneration in mice Pavle Mataka, Andrija Mataka, Sarah Moustafaa, Dipendra K. Aryalb,c,d,e, Eric J. Bennerf, William Wetselb,c,d,e, and Nancy C. Andrewsa,f,1 aDepartment of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, NC 27705; bDepartment of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, NC 27705; cDepartment of Cell Biology, Duke University School of Medicine, Durham, NC 27705; dDepartment of Neurobiology, Duke University School of Medicine, Durham, NC 27705; eMouse Behavioral and Neuroendocrine Analysis Core Facility, Duke University School of Medicine, Durham, NC 27705; and fDepartment of Pediatrics, Duke University School of Medicine, Durham, NC 27705 This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected in 2015. Contributed by Nancy C. Andrews, January 27, 2016 (sent for review September 30, 2015; reviewed by Michael K. Georgieff and Barry H. Paw) Disrupted brain iron homeostasis is a common feature of neuro- offers a very sensitive approach to detect cellular iron deficiency degenerative disease. To begin to understand how neuronal iron and surfeit (12). handling might be involved, we focused on dopaminergic neurons Iron homeostasis is altered locally, in the affected part of the and asked how inactivation of transport proteins affected iron brain, in most human neurodegenerative disorders (13). Iron ac- homeostasis in vivo in mice. Loss of the cellular iron exporter, cumulates in the substantia nigra (SN) in Parkinson’s disease (PD) ferroportin, had no apparent consequences. However, loss of (14–16), although it is unsettled whether increased iron is in neu- transferrin receptor 1, involved in iron uptake, caused neuronal ropil (17) or dopaminergic (DA) neurons (18), or both. -

Targeting Non-Oncogene Addiction for Cancer Therapy
biomolecules Review Targeting Non-Oncogene Addiction for Cancer Therapy Hae Ryung Chang 1,*,†, Eunyoung Jung 1,†, Soobin Cho 1, Young-Jun Jeon 2 and Yonghwan Kim 1,* 1 Department of Biological Sciences and Research Institute of Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea; [email protected] (E.J.); [email protected] (S.C.) 2 Department of Integrative Biotechnology, Sungkyunkwan University, Suwon 16419, Korea; [email protected] * Correspondence: [email protected] (H.R.C.); [email protected] (Y.K.); Tel.: +82-2-710-9552 (H.R.C.); +82-2-710-9552 (Y.K.) † These authors contributed equally. Abstract: While Next-Generation Sequencing (NGS) and technological advances have been useful in identifying genetic profiles of tumorigenesis, novel target proteins and various clinical biomarkers, cancer continues to be a major global health threat. DNA replication, DNA damage response (DDR) and repair, and cell cycle regulation continue to be essential systems in targeted cancer therapies. Although many genes involved in DDR are known to be tumor suppressor genes, cancer cells are often dependent and addicted to these genes, making them excellent therapeutic targets. In this review, genes implicated in DNA replication, DDR, DNA repair, cell cycle regulation are discussed with reference to peptide or small molecule inhibitors which may prove therapeutic in cancer patients. Additionally, the potential of utilizing novel synthetic lethal genes in these pathways is examined, providing possible new targets for future therapeutics. Specifically, we evaluate the potential of TONSL as a novel gene for targeted therapy. Although it is a scaffold protein with no known enzymatic activity, the strategy used for developing PCNA inhibitors can also be utilized to target TONSL. -

Plenary and Platform Abstracts
American Society of Human Genetics 68th Annual Meeting PLENARY AND PLATFORM ABSTRACTS Abstract #'s Tuesday, October 16, 5:30-6:50 pm: 4. Featured Plenary Abstract Session I Hall C #1-#4 Wednesday, October 17, 9:00-10:00 am, Concurrent Platform Session A: 6. Variant Insights from Large Population Datasets Ballroom 20A #5-#8 7. GWAS in Combined Cancer Phenotypes Ballroom 20BC #9-#12 8. Genome-wide Epigenomics and Non-coding Variants Ballroom 20D #13-#16 9. Clonal Mosaicism in Cancer, Alzheimer's Disease, and Healthy Room 6A #17-#20 Tissue 10. Genetics of Behavioral Traits and Diseases Room 6B #21-#24 11. New Frontiers in Computational Genomics Room 6C #25-#28 12. Bone and Muscle: Identifying Causal Genes Room 6D #29-#32 13. Precision Medicine Initiatives: Outcomes and Lessons Learned Room 6E #33-#36 14. Environmental Exposures in Human Traits Room 6F #37-#40 Wednesday, October 17, 4:15-5:45 pm, Concurrent Platform Session B: 24. Variant Interpretation Practices and Resources Ballroom 20A #41-#46 25. Integrated Variant Analysis in Cancer Genomics Ballroom 20BC #47-#52 26. Gene Discovery and Functional Models of Neurological Disorders Ballroom 20D #53-#58 27. Whole Exome and Whole Genome Associations Room 6A #59-#64 28. Sequencing-based Diagnostics for Newborns and Infants Room 6B #65-#70 29. Omics Studies in Alzheimer's Disease Room 6C #71-#76 30. Cardiac, Valvular, and Vascular Disorders Room 6D #77-#82 31. Natural Selection and Human Phenotypes Room 6E #83-#88 32. Genetics of Cardiometabolic Traits Room 6F #89-#94 Wednesday, October 17, 6:00-7:00 pm, Concurrent Platform Session C: 33. -
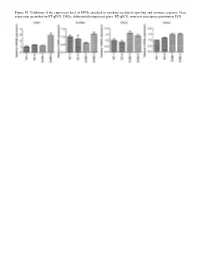
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine