Genetic Characterization and Implications for Conservation of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Systematics and Biodiversity Molecular DNA Identity of the Mouflon of Cyprus (Ovis Orientalis Ophion, Bovidae): Near Eastern
This article was downloaded by: [Università degli Studi di Milano] On: 17 August 2015, At: 02:13 Publisher: Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: 5 Howick Place, London, SW1P 1WG Systematics and Biodiversity Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/tsab20 Molecular DNA identity of the mouflon of Cyprus (Ovis orientalis ophion, Bovidae): Near Eastern origin and divergence from Western Mediterranean conspecific populations Monica Guerrinia, Giovanni Forcinaa, Panicos Panayidesb, Rita Lorenzinic, Mathieu Gareld, Petros Anayiotosb, Nikolaos Kassinisb & Filippo Barbaneraa a Dipartimento di Biologia, Unità di Zoologia e Antropologia, Via A. Volta 4, 56126 Pisa, Italy b Game Fund Service, Ministry of Interior, 1453 Nicosia, Cyprus c Istituto Zooprofilattico Sperimentale delle Regioni Lazio e Toscana, Centro di Referenza Click for updates Nazionale per la Medicina Forense Veterinaria, Via Tancia 21, 02100 Rieti, Italy d Office National de la Chasse et de la Faune Sauvage, Centre National d'Études et de Recherche Appliquée Faune de Montagne, 5 allée de Bethléem, Z.I. Mayencin, 38610 Gières, France Published online: 11 Jun 2015. To cite this article: Monica Guerrini, Giovanni Forcina, Panicos Panayides, Rita Lorenzini, Mathieu Garel, Petros Anayiotos, Nikolaos Kassinis & Filippo Barbanera (2015) Molecular DNA identity of the mouflon of Cyprus (Ovis orientalis ophion, Bovidae): Near Eastern origin and divergence from Western Mediterranean conspecific populations, Systematics and Biodiversity, 13:5, 472-483, DOI: 10.1080/14772000.2015.1046409 To link to this article: http://dx.doi.org/10.1080/14772000.2015.1046409 PLEASE SCROLL DOWN FOR ARTICLE Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. -
Cic Pheonotype List Caprinae©
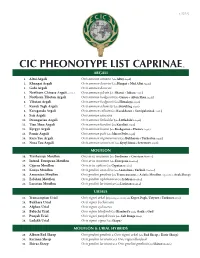
v. 5.25.12 CIC PHEONOTYPE LIST CAPRINAE © ARGALI 1. Altai Argali Ovis ammon ammon (aka Altay Argali) 2. Khangai Argali Ovis ammon darwini (aka Hangai & Mid Altai Argali) 3. Gobi Argali Ovis ammon darwini 4. Northern Chinese Argali - extinct Ovis ammon jubata (aka Shansi & Jubata Argali) 5. Northern Tibetan Argali Ovis ammon hodgsonii (aka Gansu & Altun Shan Argali) 6. Tibetan Argali Ovis ammon hodgsonii (aka Himalaya Argali) 7. Kuruk Tagh Argali Ovis ammon adametzi (aka Kuruktag Argali) 8. Karaganda Argali Ovis ammon collium (aka Kazakhstan & Semipalatinsk Argali) 9. Sair Argali Ovis ammon sairensis 10. Dzungarian Argali Ovis ammon littledalei (aka Littledale’s Argali) 11. Tian Shan Argali Ovis ammon karelini (aka Karelini Argali) 12. Kyrgyz Argali Ovis ammon humei (aka Kashgarian & Hume’s Argali) 13. Pamir Argali Ovis ammon polii (aka Marco Polo Argali) 14. Kara Tau Argali Ovis ammon nigrimontana (aka Bukharan & Turkestan Argali) 15. Nura Tau Argali Ovis ammon severtzovi (aka Kyzyl Kum & Severtzov Argali) MOUFLON 16. Tyrrhenian Mouflon Ovis aries musimon (aka Sardinian & Corsican Mouflon) 17. Introd. European Mouflon Ovis aries musimon (aka European Mouflon) 18. Cyprus Mouflon Ovis aries ophion (aka Cyprian Mouflon) 19. Konya Mouflon Ovis gmelini anatolica (aka Anatolian & Turkish Mouflon) 20. Armenian Mouflon Ovis gmelini gmelinii (aka Transcaucasus or Asiatic Mouflon, regionally as Arak Sheep) 21. Esfahan Mouflon Ovis gmelini isphahanica (aka Isfahan Mouflon) 22. Larestan Mouflon Ovis gmelini laristanica (aka Laristan Mouflon) URIALS 23. Transcaspian Urial Ovis vignei arkal (Depending on locality aka Kopet Dagh, Ustyurt & Turkmen Urial) 24. Bukhara Urial Ovis vignei bocharensis 25. Afghan Urial Ovis vignei cycloceros 26. -

Impact of Climatic Factors on the Success of Hunting Various Game Species in Czech Republic - 2989
Rösslová et al.: Impact of climatic factors on the success of hunting various game species in Czech Republic - 2989 - IMPACT OF CLIMATIC FACTORS ON THE SUCCESS OF HUNTING VARIOUS GAME SPECIES IN CZECH REPUBLIC RÖSSLOVÁ, M.1 – VACEK, S.2 – VACEK, Z.2* – PROKŮPKOVÁ, A.2 1Czech University of Life Sciences Prague, Faculty of Environmental Sciences Kamýcká 129, 165 21 Prague 6 - Suchdol, Czech Republic 2Czech University of Life Sciences Prague, Faculty of Forestry and Wood Sciences Kamýcká 129, 165 21 Prague 6 - Suchdol, Czech Republic *Corresponding author e-mail: [email protected] (Received 14th Nov 2019; accepted 12th Feb 2020) Abstract. The study evaluates the impact of climatic factors on the success of hunting in the Doupov Hills in the Czech Republic. The aim was to assess the impact of climatic factors on the success of individual and group hunting of various game species and their sex and age between 2005–2012. Overall, 27 thousand animals of the following game species were analyzed: red deer (Cervus elaphus L.), sika deer (Cervus nippon Temminck), fallow deer (Dama dama L.), mouflon (Ovis musimon L.), roe deer (Capreolus capreolus L.), wild boar (Sus scrofa L.), European badger (Meles meles L.) and red fox (Vulpes vulpes L.). The results show that mostly temperature co-determines the success of both hunting methods. Atmospheric pressure represents another important factor of influence, especially in group hunting. Snow cover supports variability of the probability of game hunting success. The success of hunting increased with decreasing temperature, increasing snow cover and increasing atmospheric pressure. Conversely, precipitation and wind speed had low effect on the success of hunting. -

157 Phenotype Evaluation of Free-Ranging European Mouflon
SHORT COMMUNICATION TAPROBANICA , ISSN 1800–427X. December, 2013. Vol. 05, No. 02: pp. 157–162. © Taprobanica Private Limited, 146, Kendalanda, Homagama, Sri Lanka. http://www.sljol.info/index.php/tapro Phenotype evaluation of free-ranging however, there have not been domestic sheep, European mouflon mouflon-domestic sheep crosses, feral goats, or feral pigs on Lanai for more than 30 years. Since introduced to Lanai in 1954 (Tomich, European mouflon will readily cross with 1986) European mouflon have been as domestic sheep and will produce fertile controversial as feral domestic sheep, sheep offspring (Tomiczek, 1985; Mitchell & Frisina, hybrids, and mouflon introduced to other 2007). Thus, the question of how “true” to the Isands in the Hawaiian Archipelago (Hess & phenotype various hunted populations are is Banko, 2011). To conservationists concerned often raised by hunters and hunter-conservation with the impacts to native flora and fauna organizations. European mouflon are an introduced ungulate pest that needs to be eliminated to avoid European Mouflon: European mouflon are degradation of the native environment and native to the Mediterranean islands of Corsica indigenous wild species (Hess & Banko, 2011). and Sardinia, and also Cyprus where a slightly On the other hand European mouflon are different phenotype occurs. Mouflon are widely considered a highly valued species for distributed due to introductions to the Crimea, recreational and trophy hunting (Frisina & Germany, Switzerland, Holland, Luxemburg, Frisina, 2005) and in some locations, like Italy, Poland, the Czech Republic, Slovakia, Lanai, are important economically in a difficult Hungary, Yugoslavia, Romania, Spain, economy. Many residents of Lanai value the Finland, Denmark, Bulgaria, and the United recreational hunting of mouflon on the 14,000 States (Clark, 1964; Uloth, 1976; Valdez, 1982; ha Lanai public game management area Mitchell & Frisina, 2007). -

Animal Board Invited Review: Sheep Birth Distribution in Past Herds: a Review for Prehistoric Europe (6Th to 3Rd Millennia BC) Marie Balasse, A
Animal Board Invited Review: Sheep birth distribution in past herds: a review for prehistoric Europe (6th to 3rd millennia BC) Marie Balasse, A. Tresset, A. Bălăşescu, E. Blaise, C. Tornero, H. Gandois, D. Fiorillo, É. Á. Nyerges, D. Frémondeau, E. Banffy, et al. To cite this version: Marie Balasse, A. Tresset, A. Bălăşescu, E. Blaise, C. Tornero, et al.. Animal Board Invited Review: Sheep birth distribution in past herds: a review for prehistoric Europe (6th to 3rd millennia BC). animal, Published by Elsevier (since 2021) / Cambridge University Press (until 2020), 2017, 11 (12), pp.2229-2236. 10.1017/S1751731117001045. hal-02113946 HAL Id: hal-02113946 https://hal.archives-ouvertes.fr/hal-02113946 Submitted on 29 Apr 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Animal, page 1 of 8 © The Animal Consortium 2017. This is an Open Access article, distributed under the terms of the Creative Commons Attribution licence (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted re-use, distribution, and reproduction in any medium, provided the original work is properly cited. animal doi:10.1017/S1751731117001045 Animal Board Invited Review: Sheep birth distribution in past herds: a review for prehistoric Europe (6th to 3rd millennia BC) M. -

The Influence of Age on the Activity of Selected Biochemical
animals Article The Influence of Age on the Activity of Selected Biochemical Parameters of the Mouflon (Ovis musimon L.) Terézia Pošiváková 1,* , Jozef Švajlenka 2,* ,Ján Pošivák 3, Jaroslav Pokorádi 4, Rudolf Hromada 1, Peter Korim 1 and Ladislav Molnár 5 1 Department of the Environment, Veterinary Legislation and Economy, University of Veterinary Medicine and Pharmacy of Košice, Komenského 73, 041 81 Košice, Slovakia; [email protected] (R.H.); [email protected] (P.K.) 2 Department of Construction Technology and Management, Faculty of Civil Engineering, Technical University of Košice, Vysokoškolská 4, 042 00 Košice, Slovakia 3 Clinic for ruminants, University of Veterinary Medicine and Pharmacy of Kosice, Komenského 73, 041 81 Košice, Slovak Republic; [email protected] 4 Xcell Breeding Services Ltd., 81101 Bratislava, Slovakia; [email protected] 5 Clinic for birds and exotic animals, University of Veterinary Medicine and Pharmacy of Kosice, Komenského 73, 041 81 Košice, Slovak Republic; [email protected] * Correspondence: [email protected] (T.P.); [email protected] (J.Š.) Received: 27 March 2019; Accepted: 9 May 2019; Published: 15 May 2019 Simple Summary: Blood analyses can be used to assess the health and physiological conditions of wild animals and may provide a precise picture of disease, habitat quality, and other environmental factors. The objective of this study was to analyze the relationship between the age and selected biochemical parameters of the female mouflon (Ovis musimon L.). This study aims to create a possible reference range of biochemical parameter concentrations in the mouflon and to extend the knowledge of this species of wild game within our geographic region. -

Genetic Variability in French Populations of the Corsican Mouflon (Ovis Ammon Musimon): Analysis of 2Blood Proteins and Red-Cell Blood Groups C
Genetic variability in french populations of the corsican mouflon (Ovis ammon musimon): analysis of 2blood proteins and red-cell blood groups C. Montgelard, T.C. Nguyen, D. Dubray To cite this version: C. Montgelard, T.C. Nguyen, D. Dubray. Genetic variability in french populations of the corsican mouflon (Ovis ammon musimon): analysis of 2 blood proteins and red-cell blood groups. Genetics Selection Evolution, BioMed Central, 1994, 26 (4), pp.303-315. hal-02710257 HAL Id: hal-02710257 https://hal.inrae.fr/hal-02710257 Submitted on 1 Jun 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Original article Genetic variability in French populations of the Corsican mouflon (Ovis ammon musimon): analysis of 2 blood proteins and red-cell blood groups C Montgelard TC Nguyen2 D Dubray 1 Institut des Sciences de l’Évolution (URA 827 CNRS), Laboratoire de Paléontologie des Vertébrés (EPHE), UM II, CC 64, 3!095 Montpellier Cedex 5; 2 INRA, Unité Polymorphisme Sanguin Ovin et Caprin, Laboratoire des Groupes Sanguins, 78350 Jouy-en-Josas; 3 Office National de la Chasse, CNERA, Faune de Montagne, BP 607,!, 84030 Montpellier Cedex 1, France (Received 8 April 1993; accepted 11 February 1994) Summary - Genetic variation in 7 French populations of the Corsican mouflon (Ovis ammon musimon) was investigated by red-cell blood-group typing and electrophoresis of hemoglobin and transferrin. -

Controlling Wild Ungulates in Hawaii
Controlling Wild Sheep and Deer on Conservation Lands in Hawai‘i Position Paper 2007-01 Hawai‘i Conservation Alliance Position Statement Wild ungulates (European mouflon, axis deer and Columbian black-tailed deer) introduced to Hawai‘i are detrimental to Hawaii’s native ecosystems via the damage they inflict on both vegetation structure and composition. These animals are very difficult to control. They have high population growth rates, are elusive, and can jump or circumvent most existing ungulate fences. Control and/or removal of these animals should be a high priority on all lands designated for protection of native biodiversity in Hawai‘i. Summary The highly endemic ecosystems of the Hawaiian Islands evolved in the absence of large land mammals and thus are vulnerable to browsing and other impacts of the numerous ungulates1, 2 introduced to Hawai‘i. These ungulates now including feral domesticated species such as cattle, pigs, and goats, and wild species such as European mouflon sheep and feral sheep hybrids, axis deer and Columbian black- tailed deer. Wild ungulates are more difficult to manage than their feral domestic relatives [See HCA Position Paper 2005.1] because they are able to jump fences that normally control feral species. However, if control measures are not implemented these wild species will expand their ranges and population numbers, severely limiting our ability to protect the endemic Hawaiian biota and ecosystems. Wild sheep and deer in Hawai‘i The European mouflon (an undomesticated form of Ovis aries l .) and Armenian mouflon (O. orientalis Gmelin) are wild sheep that are closely related to the early ancestors of domestic sheep (O. -

On the Origin of European Sheep As Revealed By
On the origin of European sheep as revealed by the diversity of the Balkan breeds and by optimizing population-genetic analysis tools Elena Ciani, Salvatore Mastrangelo, Anne da Silva, Fabio Marroni, Maja Ferenčaković, Paolo Ajmone-Marsan, Hayley Baird, Mario Barbato, Licia Colli, Chiara Delvento, et al. To cite this version: Elena Ciani, Salvatore Mastrangelo, Anne da Silva, Fabio Marroni, Maja Ferenčaković, et al.. On the origin of European sheep as revealed by the diversity of the Balkan breeds and by optimizing population-genetic analysis tools. Genetics Selection Evolution, BioMed Central, 2020, 52 (1), pp.25. 10.1186/s12711-020-00545-7. hal-02610772 HAL Id: hal-02610772 https://hal.archives-ouvertes.fr/hal-02610772 Submitted on 18 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Ciani et al. Genet Sel Evol (2020) 52:25 https://doi.org/10.1186/s12711-020-00545-7 Genetics Selection Evolution RESEARCH ARTICLE Open Access On the origin of European sheep as revealed by the diversity of the Balkan breeds and by optimizing population-genetic analysis tools Elena Ciani1, Salvatore Mastrangelo2, Anne Da Silva3, Fabio Marroni4, Maja Ferenčaković5, Paolo Ajmone‑Marsan6, Hayley Baird7, Mario Barbato6, Licia Colli6, Chiara Delvento1, Toni Dovenski8, Gregor Gorjanc9, Stephen J. -

October 2004 (Hess Et Al
Caprinae Newsletter of the IUCN/SSC Caprinae Specialist Group October 2004 (Hess et al. 1997). In Pakistan, these urial subdesert, with lower slope precipitation In this issue are now primarily confined to Chitral ranging between 100 and 200 mm a year District in the Northwest Frontier and temperatures soaring over 45° C Ladakh Urial in Pakistan ……. 1 Province and Baltistan District (Skardu during summer days. In the mountains on Wild Goat and Armenian region) in the Northern Areas. However, a either side of the Indus there are definite Mouflon in Armenia ………..… 3 small population is recorded in Hispar floral zones. Below 1,800 m is essentially Taxonomic News Valley, Nagar region of Gilgit District. barren and rocky desert with little European Mouflon.……….…. 4 Although Hess et al. (1997) found “no vegetation outside of villages with their evidence of its presence within the whole irrigated crops. Above 1,800 m, vegetation Abstracts .…....................……5 area along the Gilgit and Indus rivers is dominated by sagebrush or wormwood upstream from Gilgit to downstream from (Artemisia), while scattered oak (Quercus Editorial Chilas,” a small population has been baloot) and juniper (Juniperus macropoda) recorded in that region in Bunji Valley of appear around 2,100 m. At higher Please remember to keep submitting Diamer District (IUCN-Pakistan pers. elevations, between 2,500 to 3,800 m, there articles for your newsletter – Thank you. comm.). are often open pine forests (Pinus David Shackleton wallichiana and P. gerardiana), with Editor Location juniper and patches of deodar cedar (Cedrus Southern Gilgit and Diamer Districts deodara ) and spruce (Picea smithiana). -

Estimation of Inbreeding and Effective Population Size in Istrian Cattle
Conservation genomics in the management of the Mouflon population from the hunting area Kalifront Cubric-Curik Vlatka1, Oršanić Milan2, Ugarković Damir2, Ferenčaković Maja1, Curik Ino1 1Department of Animal Science - Faculty of Agriculture & Department of Forest 2Ecology and Silviculture - Faculty of Forestry , University of Zagreb, Croatia Kalifront - Rab: open state hunting area (13,51 km2) 1998 : 41 mouflons (33 F + 8 M) Current population size ≈ 270 mouflons (1.35 F : 1 M) Observed decline in trophy values & body weight → last 10 years Aims of this study 1. To estimate genomic level of inbreeding 2. To estimate effective populations size 3. To analyse ticks paralysis lethality by autozygosity mapping Materials & methods Data and molecular analysis - Our sample: 32 Rab mouflons OvineSNP50 BeadChip (Illumina) - Public data used: EU mouflon, Sardinian mouflon, Soay sheep, Finnsheep, Australian Merino, Churra and Sardinian White Dryad digital repository: http://dx.doi.org/10.5061/dryad.2p0qf 54,241 SNPs Genomic analysis OvineSNP50 Genotyping BeadChip Data editing and cleaning in PLINK - GT score > 0.4 - GC score > 0.7 - Mind < 0.05 - Geno < 0.1 47708 polymorphic and 2549 monomorphic loci on autosomes Estimation of genomic (ROH) inbreeding - Ferenčaković et al., 2013 GSE - Software: SVS (SNP Variation Suite). An ROH is defined as a continuous (uninterrupted) stretch of DNA sequence without heterozygosity in diploid state. ROH length (k): >4Mb, >8Mb, >16Mb, 4-8Mb FROH4-8Mb FROH16>Mb FROH8>Mb FROH>4Mb ROH (Mb) 4 8 16 E[common Ancestor] 12.5 -

Antelopes, Gazelles, Cattle, Goats, Sheep, and Relatives
© Copyright, Princeton University Press. No part of this book may be distributed, posted, or reproduced in any form by digital or mechanical means without prior written permission of the publisher. INTRODUCTION RECOGNITION The family Bovidae, which includes Antelopes, Cattle, Duikers, Gazelles, Goats, and Sheep, is the largest family within Artiodactyla and the most diverse family of ungulates, with more than 270 recent species. Their common characteristic is their unbranched, non-deciduous horns. Bovids are primarily Old World in their distribution, although a few species are found in North America. The name antelope is often used to describe many members of this family, but it is not a definable, taxonomically based term. Shape, size, and color: Bovids encompass an extremely wide size range, from the minuscule Royal Antelope and the Dik-diks, weighing as little as 2 kg and standing 25 to 35 cm at the shoulder, to the Asian Wild Water Buffalo, which weighs as much as 1,200 kg, and the Gaur, which measures up to 220 cm at the shoulder. Body shape varies from relatively small, slender-limbed, and thin-necked species such as the Gazelles to the massive, stocky wild cattle (fig. 1). The forequarters may be larger than the hind, or the reverse, as in smaller species inhabiting dense tropical forests (e.g., Duikers). There is also a great variety in body coloration, although most species are some shade of brown. It can consist of a solid shade, or a patterned pelage. Antelopes that rely on concealment to avoid predators are cryptically colored. The stripes and blotches seen on the hides of Bushbuck, Bongo, and Kudu also function as camouflage by helping to disrupt the animals’ outline.