125526Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Softraid Boot
softraid boot Stefan Sperling <[email protected]> EuroBSDcon 2015 Introduction to softraid OpenBSD's softraid(4) device emulates a host controller which provides a virtual SCSI bus uses disciplines to perform I/O on underlying disks: RAID 0, RAID 1, RAID 5, CRYPTO, CONCAT borrows the bioctl(8) configuration utility from the bio(4) hardware RAID abstraction layer softraid0 at root scsibus4 at softraid0: 256 targets sd9 at scsibus4 targ 1 lun 0: <OPENBSD, SR RAID 1, 005> SCSI2 0/direct fixed sd9: 1430796MB, 512 bytes/sector, 2930271472 sectors (RAID 1 softraid volume appearing as disk sd9) OpenBSD softraid boot 2/22 Introduction to softraid OpenBSD's softraid(4) device uses chunks (disklabel slices of type RAID) for storage records meta data at the start of each chunk: format version, UUID, volume ID, no. of chunks, chunk ID, RAID type and size, and other optional meta data # disklabel -pm sd2 [...] # size offset fstype [fsize bsize cpg] c: 1430799.4M 0 unused d: 1430796.9M 64 RAID # bioctl sd9 Volume Status Size Device softraid0 0 Online 1500298993664 sd9 RAID1 0 Online 1500298993664 0:0.0 noencl <sd2d> 1 Online 1500298993664 0:1.0 noencl <sd3d> (RAID 1 softraid volume using sd2d and sd3d for storage) OpenBSD softraid boot 3/22 Introduction to softraid softraid volumes can be assembled manually with bioctl(8) or automatically during boot softraid UUID ties volumes and chunks together disk device names and disklabel UUIDs are irrelevant when softraid volumes are auto-assembled volume IDs are used to attach volumes in a predictable order stable disk device names unless disks are added/removed chunk IDs make chunks appear in a predictable order important for e.g. -

Getting Started with Openbsd Device Driver Development
Getting started with OpenBSD device driver development Stefan Sperling <[email protected]> EuroBSDcon 2017 Clarifications (1/2) Courtesy of Jonathan Gray (jsg@): We don't want source code to firmware We want to be able to redistribute firmware freely Vendor drivers are often poorly written, we need to be able to write our own We want documentation on the hardware interfaces Source code is not documentation OpenBSD Device Drivers 2/46 Clarifications (2/2) I would like to add: Device driver code runs in our kernel Firmware code runs on a peripheral device, not in our kernel When we say \Binary Blobs" we are talking about closed-source device drivers, not firmware Firmware often runs with high privileges; use at your own risk If you do not want closed-source firmware, then don't buy and use hardware which needs it We are software developers, we do not build hardware We will not sign NDAs Please talk to hardware vendors and advocate for change; the current situation is very bad for free software OpenBSD Device Drivers 3/46 This talk is for ... somewhat competent C programmers who use OpenBSD and would like to use a device which lacks support so have an itch to scratch (enhance or write a driver) but are not sure where to start perhaps are not sure how the development process works and will research all further details themselves The idea for this talk came about during conversations I had at EuroBSDcon 2016. Common question: "How do I add support for my laptop's wifi device?" OpenBSD Device Drivers 4/46 Itches I have scratched.. -

The Title Title: Subtitle March 2007
sub title The Title Title: Subtitle March 2007 Copyright c 2006-2007 BSD Certification Group, Inc. Permission to use, copy, modify, and distribute this documentation for any purpose with or without fee is hereby granted, provided that the above copyright notice and this permission notice appear in all copies. THE DOCUMENTATION IS PROVIDED "AS IS" AND THE AUTHOR DISCLAIMS ALL WARRANTIES WITH REGARD TO THIS DOCUMENTATION INCLUDING ALL IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS. IN NO EVENT SHALL THE AUTHOR BE LIABLE FOR ANY SPECIAL, DIRECT, INDIRECT, OR CON- SEQUENTIAL DAMAGES OR ANY DAMAGES WHATSOEVER RESULTING FROM LOSS OF USE, DATA OR PROFITS, WHETHER IN AN ACTION OF CONTRACT, NEG- LIGENCE OR OTHER TORTIOUS ACTION, ARISING OUT OF OR IN CONNECTION WITH THE USE OR PERFORMANCE OF THIS DOCUMENTATION. NetBSD and pkgsrc are registered trademarks of the NetBSD Foundation, Inc. FreeBSD is a registered trademark of the FreeBSD Foundation. Contents Introduction v 1 Chapter Installing and Upgrading the OS and Software 1 1.1 Recognize the installation program used by each operating system . 2 1.2 Recognize which commands are available for upgrading the operating system 6 1.3 Understand the difference between a pre-compiled binary and compiling from source . 8 1.4 Understand when it is preferable to install a pre-compiled binary and how to doso ...................................... 9 1.5 Recognize the available methods for compiling a customized binary . 10 1.6 Determine what software is installed on a system . 10 1.7 Determine which software requires upgrading . 11 1.8 Upgrade installed software . 11 1.9 Determine which software have outstanding security advisories . -

Economia Com Openbsd + PF + CARP
Economia com OpenBSD + PF + CARP Anderson Nadal Humberto Sartini http://web.onda.com.br/nadal http://web.onda.com.br/humberto Palestrante Anderson Nadal ● Coordenador Técnico do Provedor OndaRPC ● Mantenedor do VDA-Postfix http://web.onda.com.br/nadal ● Participante dos projetos: ● GnuRadius http://www.gnu.org/software/radius/ ● Rau-Tu Redes Wan http://www.rau-tu.unicamp.br/redeswan/ Palestrante Humberto Sartini ● Analista de Segurança do Provedor OndaRPC ● Participante dos projetos: ● Rau-Tu Linux ( http://www.rau-tu.unicamp.br/linux ) ● HoneypotBR ( http://www.honeypot.com.br/ ) ● RootCheck ( http://www.ossec.net/rootcheck/ ) ● Participante do: ● IV e V Fórum Internacional de SL ● Conferência Internacional de SL ( Curitiba - 2003 ) ● Conisli (São Paulo/SP - 2004) Tópicos ● História do OpenBSD ● Evolução do OpenBSD ● Características do OpenBSD ● Filtro de Pacotes (PF) ● Common Address Redundancy Protocol (CARP) ● Quality Of Services (QoS) ● Virtual Private Network (VPN) ● Estudo de Caso ● Demonstração História do OpenBSD ● Por volta de 1970 várias licenças do Unix (AT&T) foram doadas para algumas universidades norte americanas ● Em Berkeley passaram a utilizar o Unix da AT&T, que provia o código fonte do sistema ● O único problema era a restrição que a licença impunha História do OpenBSD ● Criadores do Unix, programadores e a universidade formaram o grupo CSRG (Computer Systems Research Group) para adicionar extensões e novidades ao Unix ● Houve a adição do TCP/IP diretamente no kernel do sistema operacional, controle de processos, memória -

Rosetta Stone for Unix
Rosetta Stone for Unix This custom drawing feature now works in Mozilla/Firefox, in Opera 7.0 or better, and in IE 5 or better. Use click, shift-click, control-click, and the "set" and "clear" buttons to select the desired subset of OSs. Then select "Draw table" to redraw the window with your custom table. A Sysadmin's Unixersal Translator (ROSETTA STONE) OR What do they call that in this world? Contributions and corrections gratefully accepted. Please help us fill in the blanks. New "tasks" are welcome, too! PDF version (for best results print US legal size, landscape orientation. If your browser plug-in has trouble with the PDF, then download it and load it in stand-alone acroread). Framed version. OS versions in parentheses; e.g. 10+ means version 10 and greater; 9- means version 9 and previous. $=extra cost. (obs) = obsolete If not specified, commands are in one of the following directories: /usr/bin, /usr/sbin, /bin, /sbin Files referenced may be either commands, configuration files, or directly-referenced text files. Use the man command to read the man pages for details such as command options. Categories: hardware, firmware, devices disks kernel, boot, swap files, volumes networking security, backup software, patching, tracing, logging references TASK \ OS AIX A/UX DG/UX FreeBSD HP-UX IRIX Linux Mac OS X NCR Unix NetBSD OpenBSD Reliant SCO OpenServer Solaris SunOS 4 Tru64 Ultrix UNICOS OS / TASK (rh) = Red Hat, Fujitsu Siemens. Cray Inc., formed Mandrake, SUSE,... Derived from 4.4BSD- Forked from NetBSD in Originally called SINIX. Solaris 2.0-2.6, 7, 8, 9, 10 By IBM, with input from Apple 1988-1995. -

The Title Title: Subtitle March 2007
sub title The Title Title: Subtitle March 2007 Copyright c 2006-2007 BSD Certification Group, Inc. Permission to use, copy, modify, and distribute this documentation for any purpose with or without fee is hereby granted, provided that the above copyright notice and this permission notice appear in all copies. THE DOCUMENTATION IS PROVIDED "AS IS" AND THE AUTHOR DISCLAIMS ALL WARRANTIES WITH REGARD TO THIS DOCUMENTATION INCLUDING ALL IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS. IN NO EVENT SHALL THE AUTHOR BE LIABLE FOR ANY SPECIAL, DIRECT, INDIRECT, OR CON- SEQUENTIAL DAMAGES OR ANY DAMAGES WHATSOEVER RESULTING FROM LOSS OF USE, DATA OR PROFITS, WHETHER IN AN ACTION OF CONTRACT, NEG- LIGENCE OR OTHER TORTIOUS ACTION, ARISING OUT OF OR IN CONNECTION WITH THE USE OR PERFORMANCE OF THIS DOCUMENTATION. NetBSD and pkgsrc are registered trademarks of the NetBSD Foundation, Inc. FreeBSD is a registered trademark of the FreeBSD Foundation. Contents Introduction v 1 Chapter Installing and Upgrading the OS and Software 1 1.1 Recognize the installation program used by each operating system . 2 1.2 Recognize which commands are available for upgrading the operating system 6 1.3 Understand the difference between a pre-compiled binary and compiling from source . 8 1.4 Understand when it is preferable to install a pre-compiled binary and how to doso ...................................... 9 1.5 Recognize the available methods for compiling a customized binary . 10 1.6 Determine what software is installed on a system . 10 1.7 Determine which software requires upgrading . 11 1.8 Upgrade installed software . 11 1.9 Determine which software have outstanding security advisories . -

A Database Design for a Unit Status Reporting System
Calhoun: The NPS Institutional Archive Theses and Dissertations Thesis Collection 1987 A database design for a Unit Status Reporting System. Stebbins, Ann Jacoby http://hdl.handle.net/10945/22228 ;0L [A 95945-6002 NAVAL POSTGRADUATE SCHOOL Monterey, California THESIS A DATABASE DESIGN FOR A UNIT STATUS REPORTING SYSTEM by Ann Jacoby Stebbins March 1987 Thesis Advisor Y. K. Mortagy Approved for public release; distribution is unlimited T234392 un classified -- j '» H * as< a- Sn rV < *a ;.? REPORT DOCUMENTATION PAGE 'j REPORT SECURITY CLASSIFICATION 'b RESTRICTIVE MARKINGS unclassified 2a SECuR'Ty Classification authority 3 DISTRIBUTION/ AVAILABILITY OF REPORT Approved for public release; 2o OECLASSif-CATiON / DOWNGRADING SCHEDULE distribution is unlimited. 4 PERFORMING ORGANIZATION REPORT NuMBER(S) S MONITORING ORGANIZATION REPORT NuV3ER(Sj 6a NAME Of PERFORMING ORGANIZATION 6b OFFICE SYMBOL 7a NAME OF MONlTORiNG ORGANIZATION Postgraduate School (if applicable) Naval Postgraduate School Naval 54 6< ADDRESS [City State. andZlPCode) <b ADDRESS (Cry. Stite. and ZIP Code) Monterey, California 93943-5000 Monterey, California 93943-5000 9a NAME OF FUNDING/ SPONSORING 8b OFFICE SYMBOL 9 PROCUREMENT INSTRUMENT lOEN T'FiCATiON NUMBER ORGANIZATION (if applicable) 8c ADDRESS (City. State, and ZiPCode) 10 SOURCE OF F.jNOlNG NUMBERS PROGRAM PROJECT TASK WORK UNIT ELEMENT NO NO NO ACCESSION NO T T n iE (include Security Claudication) _ . A DATABASE DESIGN FOR A UNIT STATUS REPORTING SYSTEM '.£ PERSONA L AuTHOR(S) «.!. , Stebbms, Ann Jacoby • Id TYPE -
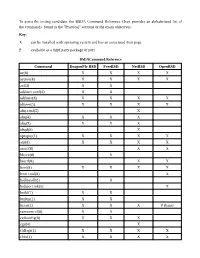
To Assist the Testing Candidate, the BSDA
To assist the testing candidate, the BSDA Command Reference Chart provides an alphabetized list of the commands found in the "Practical" sections of the exam objectives. Key: X can be installed with operating system and has an associated man page P available as a third party package or port BSDACommand Reference Command DragonFly BSD FreeBSD NetBSD OpenBSD ac(8) X X X X accton(8) X X X X acl(3) X X adduser.conf(5) X X adduser(8) X X X X aliases(5) X X X X altq.conf(5) X altq(4) X X X altq(9) X X X altqd(8) X apropos(1) X X X X arp(8) X X X X atactl(8) X X bhyve(8) X bioctl(8) X X boot(8) X X X X boot.conf(8) X bsdinstall(8) X bsd.port.mk(5) X build(7) X X builtin(1) X X bzcat(1) X X X P (bzip) camcontrol(8) X X ccdconfig(8) X X X cgd(4) X chflags(1) X X X X chfn(1) X X X X Command DragonFly BSD FreeBSD NetBSD OpenBSD chgrp(1) X X X X chmod(1) X X X X chown(8) X X X X chpass(1) X X X X chroot(8) X X X X chsh(1) X X X X cksum(1) X X X X config(8) X X X cp(1) X X X X cpdup(1) X P cpio(1) X X X X cron(8) X X X X crontab(1) X X X X crontab(5) X X X X csh(1) X X X X daily.conf(5) X daily(8) X dd(1) X X X X devinfo(8) X X df(1) X X X X dhclient.conf(5) X X X X dhclient.leases(5) X X X X dhclient(8) X X X X dhcpd.conf(5) X P X X dig(1) X X X dmesg(8) X X X X doas(1) X doas.conf(5) X domainname(1) X X X X dports(7) X drill(1) X X P du(1) X X X X dummynet(4) X X dump(8) X X X X Command DragonFly BSD FreeBSD NetBSD OpenBSD egrep(1) X X X X env(1) X X X X environ(7) X X X X etcupdate(8) X X exports(5) X X X X extattrctl(8) X X fgrep(1) X X X X file(1) -
UNISYS U 6000 Series System V Programmer's Reference Manual Volume 3 Copyright © 1988 Unisys Corporation
• UNISYS U 6000 Series System V Programmer's Reference Manual Volume 3 Copyright © 1988 Unisys Corporation. Unisys is a trademark of Unisys Corporation. March 1988 Printed in U S America Priced Item UP-13713.3 This document is intended for software releases based on AT&T Release 3 of UNIX System V or a subsequent release of the System unless otherwise indicated. NO WARRANTIES OF ANY NATURE ARE EXTENDED BY THIS DOCUMENT. Any product and related material disclosed herein are only furnished pursuant and subject to the terms and conditions of a duly executed Program Product License or Agreement to purchase or lease equipment. The only warranties made by Unisys, if any, with respect to the products described in this document are set forth in such License or Agreement. Unisys cannot accept any financial or other responsibility that may be the result of your use of the information in this document or software material, including direct, indirect, special or consequential damages. You should be very careful to ensure that the use of this information and/or software material complies with the laws, rules, and regulations of the jurisdictions with respect to which it is used. The information contained herein is subject to change without notice. Revisions may be issued to advise of such changes and/or additions. ACT, Micro-Term, and MIME are trademarks of Micro-Term. Ann Arbor is a trademark of Ann Arbor Terminals. Beehive and Superbee are registered trademarks of Beehive International. Concept is a trademark of Human Designed Systems. DEC, PDP, VAX, and VT100 are trademarks of Digital Equipment Corp. -

The BSD Associate Study Guide the BSD Associate Study Guide: the Beginning BSD Unix Administration Book
The Beginning BSD Unix Administration Book The BSD Associate Study Guide The BSD Associate Study Guide: The Beginning BSD Unix Administration Book November 24, 2011 Editor: Jeremy C. Reed Book Wiki: http://bsdwiki.reedmedia.net/ Copyright c 2006-2011 BSD Certification Group, Inc. Permission to use, copy, modify, and distribute this documentation for any purpose with or without fee is hereby granted, provided that the above copyright notice and this permission notice appear in all copies. THE DOCUMENTATION IS PROVIDED "AS IS" AND THE AUTHOR DISCLAIMS ALL WAR- RANTIES WITH REGARD TO THIS DOCUMENTATION INCLUDING ALL IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS. IN NO EVENT SHALL THE AUTHOR BE LIABLE FOR ANY SPECIAL, DIRECT, INDIRECT, OR CONSEQUENTIAL DAMAGES OR ANY DAMAGES WHATSOEVER RESULTING FROM LOSS OF USE, DATA OR PROFITS, WHETHER IN AN AC- TION OF CONTRACT, NEGLIGENCE OR OTHER TORTIOUS ACTION, ARISING OUT OF OR IN CONNECTION WITH THE USE OR PERFORMANCE OF THIS DOCUMENTATION. NetBSD and pkgsrc are registered trademarks of the NetBSD Foundation, Inc. FreeBSD is a registered trademark of the FreeBSD Foundation. Contents Introduction vii 1 Installing and Upgrading the OS and Software 1 1.1 Recognize the installation program used by each operating system . 1 1.2 Recognize which commands are available for upgrading the operating system . 5 1.3 Understand the difference between a pre-compiled binary and compiling from source . 6 1.4 Understand when it is preferable to install a pre-compiled binary and how to do so . 7 1.5 Recognize the available methods for compiling a customized binary . 7 1.6 Determine what software is installed on a system . -

Openbsd As a Full-Featured NAS
OpenBSD as a full-featured NAS OpenBSD is not only for Network related projects Fosdem 2019 Email: [email protected] Blog: https://www.vincentdelft.be My company: https://easytransitions-ict.be Topics for today ● What are the goals of this project ● How I’ve implemented it ● What were/are the problems ● Lesson’s learned ● The scripts developed are shared ● Conclusions Goals ● The goals are : – An encrypted NAS – At least 2 disks (1 for long term backup and for security) – Have a “time-machine like” system (for short term backup) – Provide files via NFS, Samba and sshfs – Every user has his own R/W folder and several other R/O folders – Delivering mp3, ogg, Flac to my hifi system + remote control it via smartphone – Deliver multi media (video, photos) to TV (~VOD) – Run on cheap HW – Easy to maintain Design TV dlna NAS FLAC JPEG/RAW MKV/AVI Working files DAC / Hifi Speakers: photo by Goh Rhy Yan on Unsplash NAS ● 3 main folders: – /mnt/sd1/share : photos, video, music. (RO) – /mnt/sd1/pfiles: personal files ● Each user has his own RW folder (not visible by others) ● Some “global folders” are RW for all users – /mnt/sd1/machines: all machine’s backups (not visible by std users) Hardware (old) ● Intel(R) Atom(TM) CPU D2500 @ 1.86GHz – Fan less – OpenBSD compatible – 4 GB Ram – 2 SATA ports – Disks 1TB Hardware ● After few years of good services, a new board with a better CPU – ASUSTeK COMPUTER INC. H110T – Fan – OpenBSD compatible – CPU 3.3 GHZ – 4GB Ram – 2 SATA ports – Same disks Lesson’s learned HW ● Read man pages -

Economia Com Openbsd + PF + CARP
Economia com OpenBSD + PF + CARP Humberto Sartini http://web.onda.com.br/humberto Palestrante Humberto Sartini ● Analista de Segurança do Provedor OndaRPC ● Participante dos projetos: ● Rau-Tu Linux ( http://www.rau-tu.unicamp.br/linux ) ● HoneypotBR ( http://www.honeypot.com.br/ ) ● RootCheck ( http://www.ossec.net/rootcheck/ ) ● Participante do: ● IV e V Fórum Internacional de SL ● Conferência Internacional de SL ( Curitiba - 2003 ) ● Conisli (São Paulo/SP – 2004) ● Latinoware (Curitiba - 2005) Tópicos ● História do OpenBSD ● Evolução do OpenBSD ● Características do OpenBSD ● Filtro de Pacotes (PF) ● Common Address Redundancy Protocol (CARP) ● Quality Of Services (QoS) ● Estudo de Caso História do OpenBSD ● Por volta de 1970 várias licenças do Unix (AT&T) foram doadas para algumas universidades norte americanas ● O único problema era a restrição que a licença impunha ● Criadores do Unix, programadores e a universidade formaram um grupo para adicionar extensões e novidades ao Unix História do OpenBSD ● Houve a adição do TCP/IP diretamente no kernel do sistema operacional, controle de processos, memória virtual, sistemas de arquivos novos e outros recursos ● Tecnicamente BSD é o nome dado aos avanços e melhorias do sistema opera- cional UNIX realizado pelo grupo História do OpenBSD ● Surgiu o grupo NetBSD para guardar, organizar, ajudar a manter o sistema, além de liberar novas releases ● O grupo FreeBSD formou-se poucos meses depois do NetBSD e tem como objetivo o suporte a arquitetura i386 História do OpenBSD ● Em meados dos anos 90,