View the System of Interactions That Occur Globally
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Outlier Detection in BLAST Hits∗
Outlier Detection in BLAST Hits∗ Nidhi Shah1, Stephen F. Altschul2, and Mihai Pop3 1 Department of Computer Science and Center for Bioinformatics and Computational Biology, University of Maryland, College Park, MD, USA [email protected] 2 Computational Biology Branch, NCBI, NLM, NIH, Bethesda, MD, USA [email protected] 3 Department of Computer Science and Center for Bioinformatics and Computational Biology, University of Maryland, College Park, MD, USA [email protected] Abstract An important task in a metagenomic analysis is the assignment of taxonomic labels to sequences in a sample. Most widely used methods for taxonomy assignment compare a sequence in the sample to a database of known sequences. Many approaches use the best BLAST hit(s) to assign the taxonomic label. However, it is known that the best BLAST hit may not always correspond to the best taxonomic match. An alternative approach involves phylogenetic methods which take into account alignments and a model of evolution in order to more accurately define the taxonomic origin of sequences. The similarity-search based methods typically run faster than phylogenetic methods and work well when the organisms in the sample are well represented in the database. On the other hand, phylogenetic methods have the capability to identify new organisms in a sample but are computationally quite expensive. We propose a two-step approach for metagenomic taxon identification; i.e., use a rapid method that accurately classifies sequences using a reference database (this is a filtering step) and then use a more complex phylogenetic method for the sequences that were unclassified in the previous step. -

Coding Sequences: a History of Sequence Comparison Algorithms As a Scientiªc Instrument
Coding Sequences: A History of Sequence Comparison Algorithms as a Scientiªc Instrument Hallam Stevens Harvard University Sequence comparison algorithms are sophisticated pieces of software that com- pare and match identical or similar regions of DNA, RNA, or protein se- quence. This paper examines the origins and development of these algorithms from the 1960s to the 1990s. By treating this software as a kind of scien- tiªc instrument used to examine sets of biological objects, the paper shows how algorithms have been used as different sorts of tools and appropriated for dif- ferent sorts of uses according to the disciplinary context in which they were deployed. These particular uses have made sequences themselves into different kinds of objects. Introduction Historians of molecular biology have paid signiªcant attention to the role of scientiªc instruments and their relationship to the production of bio- logical knowledge. For instance, Lily Kay has examined the history of electrophoresis, Boelie Elzen has analyzed the development of the ultra- centrifuge as an enabling technology for molecular biology, and Nicolas Rasmussen has examined how molecular biology was transformed by the introduction of the electron microscope (Kay 1998, 1993; Elzen 1986; Rasmussen 1997).1 Collectively, these historians have demonstrated how instruments and other elements of the material culture of the labora- tory have played a decisive role in determining the kind and quantity of knowledge that is produced by biologists. During the 1960s, a versatile new kind of instrument began to be deployed in biology: the electronic computer (Ceruzzi 2001; Lenoir 1999). Despite the signiªcant role that 1. One could also point to Robert Kohler’s (1994) work on the fruit ºy, Jean-Paul Gaudillière (2001) on laboratory mice, and Hannah Landecker (2007) on the technologies of tissue culture. -

Thesis by Submitted in Partial Fulfillment of the Requirements For
Sequential Optimization of Global Sequence Alignments Relative to Different Cost Functions Thesis by Enas Mohammad Odat, Master Degree in Computer Science Submitted in Partial Fulfillment of the Requirements for the degree of Masters of Computer Science King Abdullah University of Science and Technology Mathmatical and Computer Sciences and Engineering Division Computer System Thuwal, Makkah Province, Kingdom of Saudi Arabia May, 2011 2 The dissertation/thesis of Enas Mohammad Odat is approved by the examination committee Committee Chairperson: Committee Co-Chair: Committee Member: King Abdullah University of Science and Technology 2011 ABSTRACT Sequential Optimization of Global Sequence Alignments Relative to Different Cost Functions Enas Mohammad Odat The purpose of this dissertation is to present a methodology to model global sequence alignment problem as directed acyclic graph which helps to extract all pos- sible optimal alignments. Moreover, a mechanism to sequentially optimize sequence alignment problem relative to different cost functions is suggested. Sequence alignment is mostly important in computational biology. It is used to find evolutionary relationships between biological sequences. There are many algo- rithms that have been developed to solve this problem. The most famous algorithms are Needleman-Wunsch and Smith-Waterman that are based on dynamic program- ming. In dynamic programming, problem is divided into a set of overlapping sub- problems and then the solution of each subproblem is found. Finally, the solutions to these subproblems are combined into a final solution. In this thesis it has been proved that for two sequences of length m and n over a fixed alphabet, the suggested optimization procedure requires O(mn) arithmetic operations per cost function on a single processor machine. -

In the Last Decade Or So, the Biological Sciences Have Experience a Major
Philosophy of Bioinformatics: Extended Cognition, Analogies and Mechanisms by Joseph Mikhael A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy in Philosophy Waterloo, Ontario, Canada, 2007 ©Joseph Mikhael 2007 Author’s Declaration I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract The development of bioinformatics as an influential biological field should interest philosophers of biology and philosophers of science in general. Bioinformatics contributes significantly to the development of biological knowledge using a variety of scientific methods. Particular tools used by bioinformaticists, such as BLAST, phylogenetic tree creation software, and DNA microarrays, will be shown to utilize the scientific methods of extended cognition, analogical reasoning, and representations of mechanisms. Extended cognition is found in bioinformatics through the use of computer databases and algorithms in the representation and development of scientific theories in bioinformatics. Analogical reasoning is found in bioinformatics through particular analogical comparisons that are made between biological sequences and operations. Lastly, scientific theories that are created using certain bioinformatics tools are often representations of mechanisms. These methods are found in other scientific fields, but it will be shown that these methods are expanded in bioinformatics research through the use of computers to make the methods of analogical reasoning and representation of mechanisms more powerful. iii Acknowledgments I would like to thank first and foremost my thesis supervisor, Prof. -

BLAST) Stephen Altschul, Warren Gish, Webb Miller, Eugene Myers, David Lipman
Basic Local Alignment Search Tool (BLAST) Stephen Altschul, Warren Gish, Webb Miller, Eugene Myers, David Lipman Journal of Molecular Biology 1990 Presented for MBB752 Brian Dunican January 28, 2009 The Problem • Dynamic programming algorithms take too long when applied to large databases • Also these algorithms maximize similarity – InserQons – Deleons – Replacements • Example Needleman and Wunsch BLAST • Similarity measure is based on well defined mutaQon scores (PAM120) – OpQmizaQon of this measure approximates the results of dynamic programming algorithm • “Detect weak but biologically significant sequence similariQes, and is more than an order of magnitude faster” Methods • Matrix of Similarity – Proteins: similar +, dissimilar – (PAM120) – DNA: idenQQes +5, mismatches -4 • Units of Maximal Segment Pair (MSP) – Score can not be increased by shortening or lengthening the segment pair • BLAST seeks the highest MSP score (>=S) Method • ComputaQonally intensive to scan the database for all w-meres in search of S • Define a threshold value, T , as the lower bound for further analysis. • Database is searched for all words (w-meres) which can equal T. – Two steps • From matches with score >= T, dynamic programming is used to determine MSP – Traces out from hit to maximize score Method Matches we care about S Matches worth evaluaQng Score T Waste of Qme Parameters • The chance exists the a random sequence will exceed the S score (1) M and N are the length of the compared strings S is the arbitrary score K and lambda are coefficients Parameters • Used equaQon (1) to determine w and T parameters Time Constraints • Compile list of words that can score T from query • Scan database for matches to T-scoring words • Extend all hits to seek MSPs higher than S • Increasing w decreases Qme spent on step 3. -
![[ Team Lib ] BLAST (Basic Local Alignment Search Tool) Is a Set Of](https://docslib.b-cdn.net/cover/9460/team-lib-blast-basic-local-alignment-search-tool-is-a-set-of-7709460.webp)
[ Team Lib ] BLAST (Basic Local Alignment Search Tool) Is a Set Of
[ Team LiB ] • Table of Contents • Index • Reviews • Examples • Reader Reviews • Errata BLAST By Joseph Bedell, Ian Korf, Mark Yandell Publisher: O'Reilly Pub Date: July 2003 ISBN: 0-596-00299-8 Pages: 360 BLAST (Basic Local Alignment Search Tool) is a set of similarity search programs that explore all of the available sequence databases for protein or DNA. BLAST is the only book completely devoted to this popular and important technology and offers biologists, computational biology students, and bioinformatics professionals a clear understanding of this program. This book shows you how to get specific answers with BLAST and how to use the software to interpret results. If you have an interest in sequence analysis this is a book you should own. [ Team LiB ] [ Team LiB ] • Table of Contents • Index • Reviews • Examples • Reader Reviews • Errata BLAST By Joseph Bedell, Ian Korf, Mark Yandell Publisher: O'Reilly Pub Date: July 2003 ISBN: 0-596-00299-8 Pages: 360 Copyright Forward Preface Audience for This Book Structure of This Book A Little Math, a Little Perl Conventions Used in This Book URLs Referenced in This Book Comments and Questions Acknowledgments Part I: Introduction Chapter 1. Hello BLAST Section 1.1. What Is BLAST? Section 1.2. Using NCBI-BLAST Section 1.3. Alternate Output Formats Section 1.4. Alternate Alignment Views Section 1.5. The Next Step Section 1.6. Further Reading Part II: Theory Chapter 2. Biological Sequences Section 2.1. The Central Dogma of Molecular Biology Section 2.2. Evolution Section 2.3. Genomes and Genes Section 2.4. Biological Sequences and Similarity Section 2.5. -

Efficiency of Shared-Memory Multiprocessors for a Genetic Sequence Similarity Search Algorithm
Technical Report Department of Computer Science University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159 USA TR 97-005 Efficiency of Shared-Memory Multiprocessors for a Genetic Sequence Similarity Search Algorithm by: Ed Huai-hsin Chi, Elizabeth Shoop, John Carlis, Ernest Retzel, and John Riedl Efficiency of Shared-Memory Multiprocessors for a Genetic Sequence Similarity Search Algorithm Ed Huai-hsin Chit, Elizabeth Shoop t, John Carlist, Ernest Retzelt, John Riedlt t Computer Science Department, t Computational Biology Centers. University of Minnesota Medical School. University of Minnesota 4-192 EE/CSci Building, Box 196. UMHC. 1460 Mayo Building, Minneapolis. MN 55455 420 Delaware Street S.E., [email protected] Minneapolis, MN 55455 January 3, 1997-Version #1 Abstract Molecular biologists who conduct large-scale genetic sequencing project5 are producing an ever-increa5ing amount of sequence data. GenBank, the primary repository for DNA sequence data, is doubling in size every 1.3 years. Keeping pace with the analysis of these data is a difficult task. One of the most successful technique, for analyzing genetic data is sequence similarity analysis-U1e comparison of unknown sequences against known ,equences kept in databases. As biologists gather more sequence data, sequence similarity algorithms are more and more useful, but take longer and longer to run. BLAST is one of the most popular sequence similarity algorithms in me today, but its running time is approximately proportional to the size of the database. Sequence similarity analysis using BLAST is becoming a bottleneck in genetic sequence analysis. This paper analyzes the performance of BLAST on SMPs, to improve our theoretical and practical understanding of the scalability of the algorithm. -

Comparative Computational Analysis of Mycobacterium Species by Using Different Techniques in Study
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by International Institute for Science, Technology and Education (IISTE): E-Journals Advances in Life Science and Technology www.iiste.org ISSN 2224-7181 (Paper) ISSN 2225-062X (Online) Vol 5, 2012 Comparative Computational Analysis of Mycobacterium Species by using Different Techniques in Study * Nitesh Chandra Mishra , Kiran Singh Department of Bio-Informatics, Maulana Azad National Institute of Technology, Bhopal, M.P., India *E-mail of the Corresponding author: [email protected], [email protected] Abstract Mycobacterium tuberculosis (MTB) is a pathogenic bacteria species in the genus Mycobacterium and the causative agent of most cases of tuberculosis. It is spread through the air when people who have an active MTB infection cough, sneeze, or otherwise transmit their saliva through the air. Most infections in humans result in an asymptomatic, latent infection, and about one in ten latent infections eventually progresses to active disease, which, if left untreated, kills more than 50% of its victims. Mycobacterium tuberculosis is a member of the genus ‘tuberculosis’ in which contains various other mycobacterium species also. These species within a gene must have some similarity in them. In spite of this similarity only mycobacterium tuberculosis cause the tuberculosis disease, the remaining does not. This signifies that mycobacterium tuberculosis must be having some specific genes or proteins which are uniquely present only in it and not in the other species. This fact is used in this research and blast program is executed recursively for the comparison between these mycobacterium species. -
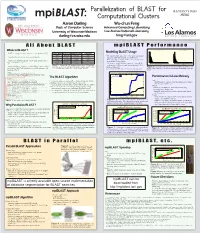
A Ll a Bout BLASTBLAST in P Arallelmpi BLASTP Erformancempi BLAST
Parallelization of BLAST for RADIANT LOGO mpiBLAST: Computational Clusters HERE Aaron Darling Wu-chun Feng Dept. of Computer Science Advanced Computing Laboratory University of Wisconsin-Madison Los Alamos National Laboratory [email protected] [email protected] All About BLAST mpiBLAST Performance What is BLAST? Predicted ORF sizes for Erwinia Chrysanthemi 3937 Length of sequence entries in the nt database Modelling BLAST Usage 1500 - BLAST is a family of sequence database search algorithms [1,2] Search Name Query Type Database Type Translation 60000 blastn Nucleotide Nucleotide None - The length, number and content of the query and database 1000 - Foundation for much research in molecular genetics tblastn Peptide Nucleotide Database 40000 Frequency sequences significantly affect BLAST runtime [11] Frequency blastx Nucleotide Peptide Query 500 - Must choose queries and the database to accurately reflect 20000 - Searches for similarities between a short query sequence and a blastp Peptide Peptide None a typical BLAST usage pattern 0 large set of database sequences. 0 tblastx Nucleotide Nucleotide Query and Database 0 5000 10000 15000 - We model a common problem in genomics: using BLAST 0 5000 10000 15000 20000 ORF length in base pairs Sequence length in base pairs Standard BLAST database search types. DNA and ami- on all predicted ORFs in a newly sequenced genome [16] - Query and database sequences are either DNA or amino acid (pro- Table 1. no acid sequences are translated by BLAST, making searches with Sequence database: nt Figure 5. Histograms of the query sequence lengths and database se- tein) sequence, BLAST will translate between DNA and amino acid several combinations of database and query type possible. -

Advances and Applications of Bioinformatics in Various Fields of Life
International Journal of Fauna and Biological Studies 2018; 5(2): 03-10 ISSN 2347-2677 IJFBS 2018; 5(2): 03-10 Advances and applications of Bioinformatics in various Received: 04-01-2018 Accepted: 05-02-2018 fields of life M Younus Wani Temperate Sericulture Research M Younus Wani, NA Ganie, S Rani, S Mehraj, MR Mir, MF Baqual, KA Institute, Mirgund, SKUAST- Kashmir, J&K, India Sahaf, FA Malik and KA Dar NA Ganie Abstract Temperate Sericulture Research Bioinformatics is an interdisciplinary area of the science composed of biology, mathematics and Institute, Mirgund, SKUAST- Kashmir, J&K, India computer science. Bioinformatics is the application of information technology to manage biological data that helps in decoding plant genomes. The field of bioinformatics emerged as a tool to facilitate S Rani biological discoveries more than 10 years ago. With the development of Human Genome Project (HGP), Temperate Sericulture Research the data of biology increased fabulously and marvelously. The ability to capture, manage, process, Institute, Mirgund, SKUAST- analyze and interpret data became more important than ever. Bioinformatics and computers can help Kashmir, J&K, India scientists to solve it. Here are introduced roles of bioinformatics, meanwhile Web tools and resources of bioinformatics are reviewed and its applications in agriculture and relevance with other disciplines is also S Mehraj highlighted. Application of various bioinformatics tools in biological research enables storage, retrieval, Temperate Sericulture Research analysis, annotation and visualization of results and promotes better understanding of biological system Institute, Mirgund, SKUAST- in fullness. This will help in animal and plant health care based disease diagnosis and treatment. -

GAME-CHANGERS Some Papers Have a Profound and Obvious Influence on Future Research and Industry Applications
INNOVATION | NATURE INDEX David Lipman, who co-authored the BLAST paper, which has been cited in at least 4,900 inventions according to patent documents in the Lens database. GAME-CHANGERS Some papers have a profound and obvious influence on future research and industry applications. Patents citing these life science papers indicate their bearing on developments which have widespread health implications. ists of the most highly cited academic patents are a general indicator of the dynamic selected from the Lens platform, based on articles garner considerable attention between science and technology, and can articles cited in patents. Each paper had been from the research community. But infer that a piece of research has influenced cited in more than 1,000 patent families by Larticles that are highly cited in patents don’t an invention (see Patently clear). Here, the 2016. Patent families represent a single inven- BILL REITZEL receive the same attention. This is surprising index profiles three life science articles that tion. Inventors often file patents in multiple given the demand from governments that sci- have been highly cited in patents. Each arti- countries, which is why the number of citing entists demonstrate the societal or economic cle has had profound impact on industry and, patents is larger than the number of patent value of their research. Citations of articles in eventually, consumers. These papers were families. NATURE INDEX 2017 | INNOVATION | S9 NATURE INDEX | INNOVATION Basic Local Alignment Search Tool (BLAST) THE GOOGLE OF GENOMES used pattern recognition software to perform ANTIBODIES AS THERAPY faster sequence comparisons. It could calculate Basic Local Alignment Search Tool the statistical level of similarity between two Replacing the complementarity- published in the Journal of Molecular sequences, says Lipman, who recently left the determining regions in a human Biology in 1990.