Major and Trace-Element Analyses of Acid Mine Waters in the Leviathan Mine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Geochemical Modeling of Iron and Aluminum Precipitation During Mixing and Neutralization of Acid Mine Drainage
minerals Article Geochemical Modeling of Iron and Aluminum Precipitation during Mixing and Neutralization of Acid Mine Drainage Darrell Kirk Nordstrom U.S. Geological Survey, Boulder, CO 80303, USA; [email protected] Received: 21 May 2020; Accepted: 14 June 2020; Published: 17 June 2020 Abstract: Geochemical modeling of precipitation reactions in the complex matrix of acid mine drainage is fundamental to understanding natural attenuation, lime treatment, and treatment procedures that separate constituents for potential reuse or recycling. The three main dissolved constituents in acid mine drainage are iron, aluminum, and sulfate. During the neutralization of acid mine drainage (AMD) by mixing with clean tributaries or by titration with a base such as sodium hydroxide or slaked lime, Ca(OH)2, iron precipitates at pH values of 2–3 if oxidized and aluminum precipitates at pH values of 4–5 and both processes buffer the pH during precipitation. Mixing processes were simulated using the ion-association model in the PHREEQC code. The results are sensitive to the solubility product constant (Ksp) used for the precipitating phases. A field example with data on discharge and water composition of AMD before and after mixing along with massive precipitation of an aluminum phase is simulated and shows that there is an optimal Ksp to give the best fit to the measured data. Best fit is defined when the predicted water composition after mixing and precipitation matches most closely the measured water chemistry. Slight adjustment to the proportion of stream discharges does not give a better fit. Keywords: geochemical modeling; acid mine drainage; iron and aluminum precipitation; schwertmannite; basaluminite 1. -

Federal Register/Vol. 67, No. 101/Friday, May 24, 2002/Notices
Federal Register / Vol. 67, No. 101 / Friday, May 24, 2002 / Notices 36645 along Monument Creek from any future Service, the California Department of and sulfur underground mining development. Fish and Game, and the Nevada operations, in Alpine County, Only one federally listed species, the Division of Environmental Protection, California. Anaconda developed the threatened Preble’s meadow jumping announces the release for public review former underground mine into an open mouse, occurs onsite and has the of the Leviathan Mine Natural Resource pit sulfur mine and operated the Mine potential to be adversely affected by the Damage Assessment Plan—Public through 1962. Anaconda sold the Mine project. To mitigate impacts that may Release Draft (Assessment Plan). The in early 1963, but no further mining result from incidental take, the HCP Plan was developed by the Leviathan operations took place thereafter. provides mitigation for the residential Mine Council Natural Resource Releases of hazardous substances site by protection of the Monument Trustees, consisting of representatives of from the Mine began in the 1950s and Creek corridor onsite and its associated the Tribe and agencies listed above, to continue today. Infiltration of riparian areas from all future assess injuries to natural resources precipitation into and through the adits development through the enhancement resulting from releases of hazardous (tunnels from the former underground of 0.5 acre through native grass planting, substances from the Leviathan Mine in mine), open pit, and overburden piles, shrub planting, weed control, Alpine County, California. The along with direct contact of mine wastes preservation in a native and unmowed Assessment Plan describes the proposed with surface waters, has created acid condition, and the placement of the approach for determining and mine drainage (AMD), which has been proposed building site closer to the road quantifying natural resource injuries released, and continues to be released and farther away from mouse habitat. -

Mining's Toxic Legacy
Mining’s Toxic Legacy An Initiative to Address Mining Toxins in the Sierra Nevada Acknowledgements _____________________________ ______________________________________________________________________________________________________________ The Sierra Fund would like to thank Dr. Carrie Monohan, contributing author of this report, and Kyle Leach, lead technical advisor. Thanks as well to Dr. William M. Murphy, Dr. Dave Brown, and Professor Becky Damazo, RN, of California State University, Chico for their research into the human and environmental impacts of mining toxins, and to the graduate students who assisted them: Lowren C. McAmis and Melinda Montano, Gina Grayson, James Guichard, and Yvette Irons. Thanks to Malaika Bishop and Roberto Garcia for their hard work to engage community partners in this effort, and Terry Lowe and Anna Reynolds Trabucco for their editorial expertise. For production of this report we recognize Elizabeth “Izzy” Martin of The Sierra Fund for conceiving of and coordinating the overall Initiative and writing substantial portions of the document, Kerry Morse for editing, and Emily Rivenes for design and formatting. Many others were vital to the development of the report, especially the members of our Gold Ribbon Panel and our Government Science and Policy Advisors. We also thank the Rose Foundation for Communities and the Environment and The Abandoned Mine Alliance who provided funding to pay for a portion of the expenses in printing this report. Special thanks to Rebecca Solnit, whose article “Winged Mercury and -

Davies Unr 0139M 11920.Pdf
University of Nevada, Reno Applications of multi-season hyperspectral remote sensing for acid mine water characterization and mapping of secondary iron minerals associated with acid mine drainage A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Hydrogeology by Gwendolyn E. Davies Dr. Wendy M. Calvin/Thesis Advisor December, 2015 © by Gwendolyn E. Davies 2015 All Rights Reserved THE GRADUATE SCHOOL We recommend that the thesis prepared under our supervision by GWENDOLYN E. DAVIES entitled Applications of Multi-Season Hyperspectral Remote Sensing for Acid Mine Water Characterization and Mapping of Secondary Iron Minerals Associated with Acid Mine Drainage be accepted in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Wendy M. Calvin, Ph.D., Advisor Simon R. Poulson, Ph.D., Committee Member Glenn C. Miller, Ph.D., Graduate School Representative David W. Zeh, Ph.D., Dean, Graduate School December, 2015 i Abstract: Acid mine drainage (AMD) resulting from the oxidation of sulfides in mine waste is a major environmental issue facing the mining industry today. Open pit mines, tailings ponds, ore stockpiles, and waste rock dumps can all be significant sources of pollution, primarily heavy metals. These large mining-induced footprints are often located across vast geographic expanses and are difficult to access. With the continuing advancement of imaging satellites, remote sensing may provide a useful monitoring tool for pit lake water quality and the rapid assessment of abandoned mine sites. This study explored the applications of laboratory spectroscopy and multi-season hyperspectral remote sensing for environmental monitoring of mine waste environments. -

5.1 Historic Period Human Interaction with the Watershed
Upper Carson River Watershed Stream Corridor Assessment 5. Human Interaction With the Watershed 5.1 Historic Period Human Interaction With the Watershed The purpose of this section is to summarize human activities that have had some effect on the Carson River watershed in Alpine County, California. Regional prehistory and ethnography are summarized by Nevers (1976), Elston (1982), d’Azevedo (1986), and Lindstrom et al. (2000). Details of regional history can be found in Maule (1938), Jackson (1964), Dangberg (1972), Clark (1977), Murphy (1982), Marvin (1997), and other sources. A book published by the Centennial Book Committee (1987) contains an excellent selection of historic photographs. Particularly useful is a study on the historical geography of Alpine County by Howatt (1968). 5.1.1 Prehistoric Land Use Human habitation of the Upper Carson River Watershed extends thousands of years back into antiquity. Archaeological evidence suggests use of the area over at least the last 8,000 to 9,000 years. For most of that time, the land was home to small bands of Native Americans. Their number varied over time, depending on regional environmental conditions. For at least the last 2,000 years, the Washoe occupied the Upper Carson River Watershed. Ethnographic data provides clues as to past land use and land management practices (see extended discussions in Downs 1966; Blackburn and Anderson 1993; Lindstrom et al. 2000; Rucks 2002). A broad range of aboriginal harvesting and hunting practices, fishing, and camp tending would have affected the landscape and ecology of the study area. Shrubs such as service berry and willow were pruned to enhance growth. -

REFERENCE GUIDE to Treatment Technologies for Mining-Influenced Water
REFERENCE GUIDE to Treatment Technologies for Mining-Influenced Water March 2014 U.S. Environmental Protection Agency Office of Superfund Remediation and Technology Innovation EPA 542-R-14-001 Contents Contents .......................................................................................................................................... 2 Acronyms and Abbreviations ......................................................................................................... 5 Notice and Disclaimer..................................................................................................................... 7 Introduction ..................................................................................................................................... 8 Methodology ................................................................................................................................... 9 Passive Technologies Technology: Anoxic Limestone Drains ........................................................................................ 11 Technology: Successive Alkalinity Producing Systems (SAPS).................................................. 16 Technology: Aluminator© ............................................................................................................ 19 Technology: Constructed Wetlands .............................................................................................. 23 Technology: Biochemical Reactors ............................................................................................. -

EPA-Management and Treatment of Water from Hard Rock Mines
Management and Treatment of Water from Hard Rock Mines 1.0 PURPOSE Index The U.S. Environmental Protection Agency (EPA) Engineering Issues 1.0 PURPOSE are a new series of technology transfer documents that summarize the 2.0 SUMMARY latest available information on selected treatment and site remediation technologies and related issues. They are designed to help remedial 3.0 INTRODUCTION project managers (RPMs), on-scene coordinators (OSCs), contractors, 3.1 Background: Environmental Problems and other site managers understand the type of data and site charac at Hard-Rock Mines teristics needed to evaluate a technology for potential applicability to 3.2 Conceptual Models at Hard-Rock their specific sites. Each Engineering Issue document is developed in Mines conjunction with a small group of scientists inside the EPA and with 3.3 The Process of Selecting Remedial outside consultants, and relies on peer-reviewed literature, EPA re Technologies ports, Internet sources, current research, and other pertinent informa 3.4 Resources for Additional Information tion. The purpose of this document is to present the “state of the sci ence” regarding management and treatment of hard-rock mines. 4.0 TECHNOLOGY DESCRIPTIONS 4.1 Source Control Internet links are provided for readers interested in additional infor 4.1.1 Capping and Revegetation for mation; these Internet links, verifi ed as accurate at the time of publi Source Control cation, are subject to change. 4.1.2 Plugging Drainage Sources and Interception of Drainage by Diversion Wells 2.0 SUMMARY 4.1.3 Prevention of Acid Drainage via Contaminated water draining from hard rock mine sites continues Protective Neutralization to be a water quality problem in many parts of the U.S. -

California's Abandoned Mines
California’s Abandoned Mines A Report on the Magnitude and Scope of the Issue in the State Volume I Department of Conservation Office of Mine Reclamation Abandoned Mine Lands Unit June, 2000 TABLE OF CONTENTS: VOLUME I ACKNOWLEDGEMENTS .................................................................................................... 5 PREPARERS OF THIS REPORT ....................................................................................... 6 EXECUTIVE SUMMARY .................................................................................................... 7 OVERVIEW.......................................................................................................................... 7 KEY FINDINGS .................................................................................................................... 8 OTHER STATE AND FEDERAL AML PROGRAMS ...................................................................... 8 OPTIONS............................................................................................................................. 8 BACKGROUND .................................................................................................................11 CALIFORNIA'S MINING HISTORY .......................................................................................... 12 Metallic Mining........................................................................................................... 14 Non-Metallic Mining.................................................................................................. -

Two Bcrs and an APC at the Standard Mine Superfund Site
MULTIPLE SITE EVALUATION OF RCTS™ ACID MINE DRAINAGE TREATMENT, EMERGENCY MOBILIZATION AND LIME UTILIZATION1 2 Timothy Tsukamoto and Vance Weems Abstract. Thorough oxidation and mixing is required for treatment of acid mine drainage on many sites. This is accomplished in the rotating cylinder treatment system (RCTS™) by passing acid mine drainage and a neutralizing agent through a containment cell in which a perforated cylinder rotates. As the cylinder rotates, a thin film of water adheres to the inner and outer surfaces and water bridges across the perforations for additional gas exchange. The agitation is provided primarily by the impact of the perforations with the water flowing through the containment cell. The turbulence that is produced provides efficient mixing, which reduces chemical consumption due to more efficient use of the available alkalinity, and less sludge produced. Metals removal effectiveness, energy requirements, and chemical consumption were characterized in four field tests. In all of these, the RCTS™ effectively precipitated metals and increased pH and did so at a lower cost than conventional systems. At sites that compared the RCTS™ with conventional treatment, the RCTS™ system required substantially less energy, chemical, labor and residence time. A direct comparison with a conventional system at the Leviathan Mine demonstrated that the RCTS system used 69% less power for aeration and mixing and was more effective at oxidizing metals. The system used 41% less lime to achieve a similar discharge pH. In addition, the RCTS™ systems can be mobilized quickly to remote locations where conventional systems cannot easily be installed. System installation time was typically less than one day. -
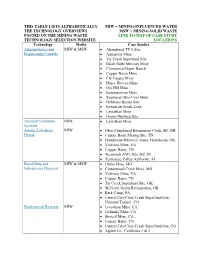
This Table Lists Alphabetically The
THIS TABLE LISTS ALPHABETICALLY MIW = MINING-INFLUENCED WATER THE TECHNOLOGY OVERVIEWS MSW = MINING SOLID WASTE POSTED ON THE MINING WASTE LINK TO MAP OF CASE STUDY TECHNOLOGY SELECTION WEBSITE LOCATIONS Technology Media Case Studies Administrative and MIW & MSW Abandoned TVA Site Engineering Controls Annapolis Mine Tar Creek Superfund Site Black Butte Mercury Mine Commerce/Mayer Ranch Copper Basin Mine Ely Copper Mine Horse Heaven Mine Ore Hill Mine Kerramerican Mine Southeast Ohio Coal Mine Gribbons Basins Site Kennecott South Zone Leviathan Mine Orono-Dunweg Site Aeration Treatment MIW Leviathan Mine Systems Anoxic Limestone MIW Ohio Abandoned Bituminous Coals, SE, OH Drains Copper Basin Mining Site, TN Hartshorne/Whitlock-Jones, Hartshorne, OK Valzinco Mine, VA Copper Basin, TN Tecumseh AML Site 262, IN Tennessee Valley Authority, AL Backfilling and MIW & MSW Hume Mine, MO Subaqueous Disposal Cottonwood Creek Mine, MO Valzinco Mine, VA Copper Basin, TN Tar Creek Superfund Site, OK McNeely Green Reclamation, OK Bark Camp, PA Central City/Clear Creek Superfund Site, National Tunnel , CO Biochemical Reactors MIW Leviathan Mine, CA Golinsky Mine, CA Stowell Mine, CA Copper Basin, TN Central City/Clear Creek Superfund Site, CO Alpine Co., California 1 & 2 Capping, Covers, and MSW Annapolis Lead Mine, MO Grading Big River mine Site, MO Dunka Mine, MN Gribbons Basin, MT Horse Heaven, OR Iron Mountain Mine, CA Kerramerican, ME Magmont Mine Site, MO Orono-Dunweg, Mo Valzinco Mine, VA Copper Basin, -

Addressing Administrative Challenges to Abandoned Mine Site Cleanup
Addressing Administrative Challenges to Abandoned Mine Site Cleanup Stephen McCord, Ph.D., P.E. Gregory J. Reller, P.G. California’s Mining Legacy in the Mountains and Valleys THE SITUATION 2 2001 Report to Legislature ~47,000 abandoned mines in California: 67% on federal land (BLM, NPS, USFS…) 2% on state/local land 31% on private land 3 Mercury Mine Site – Abbott Mine, 1949 Processing Equipment Tailings Pile Source: California Geological Survey 4 Sierra Nevada Hydraulic Mining 5 Hard Rock Gold Mine and Mill Waste Rock Stamp Mill Tailings 6 Technical Challenges to Addressing the Mining Legacy TECHNICAL CHALLENGES 7 Technical Challenges Location Hazards Resources Objectives Malakoff Diggins, 2011 8 Technical Challenge – Location Remote No Utilities Limited Access Poor Site Controls 9 Technical Challenges – Hazards Hazards Physical Chemical Metals (As, Cu, Hg, Pb, Zn…) Cyanide Asbestos 10 Technical Challenges – Resources $100,000s to $10,000,000s Many on federal or state lands Some on private lands Abbott-Turkey Run Mine Cleanup, 2006-2007 11 Technical Challenges – Objectives Protect human health Protect ecological health Protect water quality Re-use (Restoration) 12 The Laws and Regulatory Programs that Drive (or Hinder) Progress KEY STATUTES AND PROGRAMS 13 Regulatory Processes that could be applied to California AML Risk/Hazard based CERCLA California Health and Safety Code Water Quality based CWA Porter Cologne 14 Key Statutes Federal CERCLA (a.k.a. Superfund) Clean Water Act California Public Health -

Technical Evaluation of the Gold King Mine Incident
Technical Evaluation of the Gold King Mine Incident San Juan County, Colorado U.S. Department of the Interior Bureau of Reclamation Technical Service Center Denver, Colorado October 2015 Mission Statements The Department of the Interior protects and manages the Nation's natural resources and cultural heritage; provides scientific and other information about those resources; and honors its trust responsibilities or special commitments to American Indians, Alaska Natives, and affiliated island communities. The mission of the Bureau of Reclamation is to manage, develop, and protect water and related resources in an environmentally and economically sound manner in the interest of the American public. Technical Evaluation of the Gold King Mine Incident San Juan County, Colorado Peer reviewed by: U.S. Geological Survey U.S. Army Corps of Engineers U.S. Department of the Interior Bureau of Reclamation Technical Service Center Denver, Colorado October 2015 Technical Approval The results, findings, and recommendations provided in this Technical Evaluation of the Gold King Mine Incident are technically sound and consistent with current professional practice. Technical Evaluation of the Gold King Mine Incident Table of Contents page Executive Summary ...............................................................................................1 Introduction............................................................................................................5 Composition of the Evaluation Team ..................................................................6