The X Chromosome Shows Less Genetic Variation at Restriction Sites Than the Autosomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Economic Impact and Functional Applications of Human Genetics and Genomics
The Economic Impact and Functional Applications of Human Genetics and Genomics Commissioned by the American Society of Human Genetics Produced by TEConomy Partners, LLC. Report Authors: Simon Tripp and Martin Grueber May 2021 TEConomy Partners, LLC (TEConomy) endeavors at all times to produce work of the highest quality, consistent with our contract commitments. However, because of the research and/or experimental nature of this work, the client undertakes the sole responsibility for the consequence of any use or misuse of, or inability to use, any information or result obtained from TEConomy, and TEConomy, its partners, or employees have no legal liability for the accuracy, adequacy, or efficacy thereof. Acknowledgements ASHG and the project authors wish to thank the following organizations for their generous support of this study. Invitae Corporation, San Francisco, CA Regeneron Pharmaceuticals, Inc., Tarrytown, NY The project authors express their sincere appreciation to the following indi- viduals who provided their advice and input to this project. ASHG Government and Public Advocacy Committee Lynn B. Jorde, PhD ASHG Government and Public Advocacy Committee (GPAC) Chair, President (2011) Professor and Chair of Human Genetics George and Dolores Eccles Institute of Human Genetics University of Utah School of Medicine Katrina Goddard, PhD ASHG GPAC Incoming Chair, Board of Directors (2018-2020) Distinguished Investigator, Associate Director, Science Programs Kaiser Permanente Northwest Melinda Aldrich, PhD, MPH Associate Professor, Department of Medicine, Division of Genetic Medicine Vanderbilt University Medical Center Wendy Chung, MD, PhD Professor of Pediatrics in Medicine and Director, Clinical Cancer Genetics Columbia University Mira Irons, MD Chief Health and Science Officer American Medical Association Peng Jin, PhD Professor and Chair, Department of Human Genetics Emory University Allison McCague, PhD Science Policy Analyst, Policy and Program Analysis Branch National Human Genome Research Institute Rebecca Meyer-Schuman, MS Human Genetics Ph.D. -

The Human Genome Project
TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 Contents FOREWORD . 2 THE GENOME PROJECT—WHY THE DOE? . 4 A bold but logical step INTRODUCING THE HUMAN GENOME . 6 The recipe for life Some definitions . 6 A plan of action . 8 EXPLORING THE GENOMIC LANDSCAPE . 10 Mapping the terrain Two giant steps: Chromosomes 16 and 19 . 12 Getting down to details: Sequencing the genome . 16 Shotguns and transposons . 20 How good is good enough? . 26 Sidebar: Tools of the Trade . 17 Sidebar: The Mighty Mouse . 24 BEYOND BIOLOGY . 27 Instrumentation and informatics Smaller is better—And other developments . 27 Dealing with the data . 30 ETHICAL, LEGAL, AND SOCIAL IMPLICATIONS . 32 An essential dimension of genome research Foreword T THE END OF THE ROAD in Little has been rapid, and it is now generally agreed Cottonwood Canyon, near Salt that this international project will produce Lake City, Alta is a place of the complete sequence of the human genome near-mythic renown among by the year 2005. A skiers. In time it may well And what is more important, the value assume similar status among molecular of the project also appears beyond doubt. geneticists. In December 1984, a conference Genome research is revolutionizing biology there, co-sponsored by the U.S. Department and biotechnology, and providing a vital of Energy, pondered a single question: Does thrust to the increasingly broad scope of the modern DNA research offer a way of detect- biological sciences. -

Micrornas in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine
cells Review MicroRNAs in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine Danyel Fernandes Contiliani 1,2, Yasmin de Araújo Ribeiro 1,2, Vitor Nolasco de Moraes 1,2 and Tiago Campos Pereira 1,2,* 1 Graduate Program of Genetics, Department of Genetics, Faculty of Medicine of Ribeirao Preto, University of Sao Paulo, Av. Bandeirantes, Ribeirao Preto 3900, Brazil; [email protected] (D.F.C.); [email protected] (Y.d.A.R.); [email protected] (V.N.d.M.) 2 Department of Biology, Faculty of Philosophy, Sciences and Letters, University of Sao Paulo, Av. Bandeirantes, Ribeirao Preto 3900, Brazil * Correspondence: [email protected]; Tel.: +55-16-3315-3818 Abstract: MicroRNAs (miRNAs) are small non-coding RNA molecules able to post-transcriptionally regulate gene expression via base-pairing with partially complementary sequences of target tran- scripts. Prion diseases comprise a singular group of neurodegenerative conditions caused by endoge- nous, misfolded pathogenic (prion) proteins, associated with molecular aggregates. In humans, classical prion diseases include Creutzfeldt–Jakob disease, fatal familial insomnia, Gerstmann– Sträussler–Scheinker syndrome, and kuru. The aim of this review is to present the connections between miRNAs and prions, exploring how the interaction of both molecular actors may help understand the susceptibility, onset, progression, and pathological findings typical of such disorders, as well as the interface with some prion-like disorders, such as Alzheimer’s. Additionally, due to the inter-regulation of prions and miRNAs in health and disease, potential biomarkers for non-invasive miRNA-based diagnostics, as well as possible miRNA-based therapies to restore the levels of dereg- Citation: Contiliani, D.F.; Ribeiro, Y.d.A.; de Moraes, V.N.; Pereira, T.C. -

Cutting Eugenics out of CRISPR-Cas9
Ethics in Biology, Engineering & Medicine - An International Journal, 6(3–4): 263–279 (2015) Cutting Eugenics Out of CRISPR-Cas9 Carolyn Brokowski,a,* Marya Pollack,b & Robert Pollackc aBioethics (Medical Ethics) Department, Columbia University School of Professional Studies, New York, New York; bDepartment of Psychiatry, Columbia University College of Physicians and Surgeons, Inwood Clinic, New York, New York; cBiological Sciences Department, Columbia University School of the Arts, New York, New York *Address all correspondence to: Carolyn Brokowski, M.S. Candidate; Bioethics (Medical Ethics) Department, Columbia University School of Professional Studies, 203 Lewisohn Hall, 2970 Broadway, MC 4119, New York, NY 10027; E-mail: [email protected] ABSTRACT: The use of clustered regularly interspaced short palindromic repeats (CRISPR) and their associated (Cas) proteins (the CRISPR-Cas system) in genomic engineering is among the most promising biomedical innovations to occur in the last few decades. One of this system’s most profound features is its ability to edit genomes with impressive specificity, which may cause significant alterations of cellular, tissue, and organismal phenotypes at the near instance of the editing, over the lifespan of the organism and potentially into any number of future genera- tions. We argue that the use of the CRISPR-Cas9 system to edit the human germline should be legally prohibited on account of the system’s potential for generating an unjust eugenic future. Its use in nongermline experimentation and applications, however, should not be constrained on eugenic grounds. Such a blanket legal prohibition might limit the progress gleaned from this technology. Allowing experimentation in human subjects more broadly might expose par- ticipants to considerable risk and potentially harmful outcomes, and the system might prove unable to realize tangible therapeutic outcomes that seem likely ex ante. -

The Human Genome Project: Frequently Asked Questions
The Human Genome Project: Frequently Asked Questions On April 14, 2003, the National Human Genome Research Institute (NHGRI), the Department of Energy (DOE) and their partners in the International Human Genome Sequencing Consortium announced the successful completion of the Human Genome Project. What is a genome? A genome is an organism's complete set of deoxyribonucleic acid (DNA), a chemical compound that contains the genetic instructions needed to develop and direct the biological activities of every organism. DNA molecules are made of two twisting, paired strands. Each strand is made of four chemical units, called nucleotide bases. The bases are adenine (A), thymine (T), guanine (G) and cytosine (C). Bases on opposite strands pair specifically: an A always pairs with a T, and a C always with a G. The human genome contains approximately 3 billion of these base pairs, which reside in the 23 pairs of chromosomes within the nucleus of all our cells. The Human Genome’s Project’s principal task was determining the order or sequence of those As, Ts, Cs, and Gs. What is sequencing and how do you sequence a genome? Sequencing means determining the exact order of the base pairs in a segment of DNA. Human chromosomes range in size from about 50 million to 300 million base pairs. One complete set of genomic instructions is about 3 billion base pairs. Specialized machines called DNA sequencers – the technologies have rapidly evolved over the last decade – use techniques of biochemistry to automatically determine the base pair order and deposit the read out in a computer. -

The Human Genome Project and Eugenics
The Linacre Quarterly Volume 65 | Number 2 Article 6 May 1998 The umH an Genome Project and Eugenics Eduardo Rodriguez Follow this and additional works at: http://epublications.marquette.edu/lnq Recommended Citation Rodriguez, Eduardo (1998) "The umH an Genome Project and Eugenics," The Linacre Quarterly: Vol. 65: No. 2, Article 6. Available at: http://epublications.marquette.edu/lnq/vol65/iss2/6 The Human Genome Project and Eugenics by The Rev. Eduardo Rodriguez, Ph.D., M.ID. The author is Campus Minister and teaches biology at Bronx Community College. The Hwnan Genome Project is expected to provide information in the near future on the thousands of mutations that are responsible for inherited diseases, making possible the development of highly accurate genetic tests for diagnosis. On the other hand, therapeutical approaches to treat inherited diseases are not expected to develop at the same pace. In the absence oflow-cost "cures" for those born with a genetic disease it is less expensive to avoid the birth of a fetus prenatally diagnosed as having such a condition. In a society driven by economical constraints, considerable pressure on parents to abort defective children is expected. As a result, it is a matter of concern that we are arriving to a kind of society that practices a new kind of eugenics in which the individuals take the decisions and the technological advancements provide the basis for eugenic goals to be achieved without the necessity of social control. The Human Genome Project The international effort of the human genome project, which seeks to map and sequence all of the estimated 3 billion bp that make up the human genome, is expected to provide a better understanding both of single gene defects and multifactorial or familial diseases, such as diabetes and cancer. -

Human Chromosome-Specific Cdna Libraries: New Tools for Gene Identification and Genome Annotation
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press RESEARCH Human Chromosome-specific cDNA Libraries: New Tools for Gene Identification and Genome Annotation Richard G. Del Mastro, 1'2 Luping Wang, ~'2 Andrew D. Simmons, Teresa D. Gallardo, 1 Gregory A. Clines, ~ Jennifer A. Ashley, 1 Cynthia J. Hilliard, 3 John J. Wasmuth, 3 John D. McPherson, 3 and Michael Lovett ~'4 1Department of Biochemistry and the McDermott Center for Human Growth and Development, The University of Texas Southwestern Medical Center, Dallas, Texas 75235-8591; 3Department of Biological Chemistry and the Human Genome Center, University of California, Irvine, California 9271 7 To date, only a small percentage of human genes have been cloned and mapped. To facilitate more rapid gene mapping and disease gene isolation, chromosome S-specific cDNA libraries have been constructed from five sources. DNA sequencing and regional mapping of 205 unique cDNAs indicates that 25 are from known chromosome S genes and 138 are from new chromosome S genes (a frequency of 79.5%}. Sequence complexity estimates indicate that each library contains -20% of the -SO00 genes that are believed to reside on chromosome 5. This study more than doubles the number of genes mapped to chromosome S and describes an important new tool for disease gene isolation. A detailed map of expressed sequences within the pressed Sequence Tags (eSTs)] (Adams et al. 1991, human genome would provide an indispensable 1992, 1993a,b; Khan et al. 1991; Wilcox et al. resource for isolating disease genes, and would 1991; Okubo et al. -

Chromosome-Centric View of Genome Organization and Evolution
G C A T T A C G G C A T genes Editorial Chromosome-Centric View of Genome Organization and Evolution Maria Sharakhova 1,* and Vladimir Trifonov 2,* 1 Department of Entomology and Fralin Life Sciences Institute, Virginia Tech, Blacksburg, VA 24061, USA 2 Department of Genome Diversity and Evolution, Institute of Molecular and Cellular Biology, SB RAS, 630090 Novosibirsk, Russia * Correspondence: [email protected] (M.S.); [email protected] (V.T.) Genetic material in all cellular organisms is packed into chromosomes, which repre- sent essential units of inheritance, recombination, and evolution. Thus, our understanding of genome function is incomplete without knowledge of genome organization at the chro- mosome level. Recent advances in genome technologies, including long-read sequencing, chromosome flow sorting, Hi-C scaffolding, and optical mapping, allow the procurement of genome assemblies at the level of complete chromosomes. Physical genome mapping based on fluorescence in situ hybridization (FISH) enables direct correspondence of genomic sequences with particular chromosome bands or specific chromosome structures. Such chromosome-scale genome assemblies provide new opportunities for both cytogenetic and genome research. This Special Issue, entitled “Chromosome-Centric View of Genome Organization and Evolution” focuses on modern cytogenetic studies based on utilization of genome assemblies that are currently available for multiple organisms, including a key model organism, Drosophila melanogaster, and non-model organisms of vertebrates, insects, plants, and finally, humans. The issue contains twelve original research articles and 3 review papers devoted to recent achievements in the field of genomic studies of Citation: Sharakhova, M.; Trifonov, eukaryotic chromosomes. V. Chromosome-Centric View of The classical model organism D. -
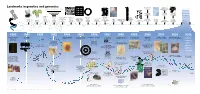
Landmarks in Genetics and Genomics
0 ++- ---+ BC PR • --- -+-t--.--+------ o,ou 1J.T33.? n.e -- -- - 4 Landmarks in genetics and genomics !!!!! U C AG +++j/GA T C US National Research U Phe Tyr Cys International Nucleotide Ser C Council issues report on U Leu stop stop A Sequence Database - stop Trp - G Mapping and Sequencing the U Consortium formed Leu Pro His Arg C Human Genome 0 5' C Gln A 3' G Human U 5' lle Asn Ser STS Markers Thr C A Met Lys Arg A G Muscular-dystrophy Development New strands Sequence-tagged Asp U of yeast artificial Genome G Val Ala Gly C gene identified sites (STS) mapping Glu A chromosome (YAC) G by positional cloning concept established cloning Archibald Garrod Project formulates Alfred Henry Oswald Avery, Colin MacLeod James Watson and Stanley Cohen and Frederick Sanger, First public Desired fragment the concept Sturtevant and Maclyn McCarty Francis Crick Marshall Nirenberg, Herbert Boyer The Belmont Report Allan Maxam discussion strands Gregor Mendel of human makes demonstrate that DNA describe the Har Gobind Khorana and develop on the use of and Walter Gilbert GenBank First human disease gene — for of sequencing First automated First-generation Cystic-fibrosis discovers Rediscovery of inborn errors the first linear is the double-helical Robert Holley determine recombinant human subjects develop DNA-sequencing database Huntington’s disease — is mapped the human The polymerase DNA-sequencing instrument human genetic Human Genome gene identified by laws of genetics Mendel’s work of metabolism map of genes hereditary material structure of DNA the genetic code DNA technology in research is issued methods established with DNA markers genome chain reaction (PCR) is invented developed map developed Organization (HUGO) formed positional cloning 1865 1900 1905 1913 1944 1953 1966 1972 1974 1977 1982 1983 1984 1985 1986 1987 1988 1989 1990 2003 +-+-o-----+---+ 1 I~I "-.■ u ''\. -

Self-Reported Sexual Behavioral Interests and Polymorphisms in the Dopamine Receptor D4 (DRD4) Exon III VNTR in Heterosexual Young Adults
Arch Sex Behav (2016) 45:2091–2100 DOI 10.1007/s10508-015-0646-6 ORIGINAL PAPER Self-Reported Sexual Behavioral Interests and Polymorphisms in the Dopamine Receptor D4 (DRD4) Exon III VNTR in Heterosexual Young Adults 1 2 3 3 Andrew C. Halley • Melanie Boretsky • David A. Puts • Mark Shriver Received: 22 January 2014 / Revised: 6 October 2015 / Accepted: 10 October 2015 / Published online: 18 November 2015 Ó Springer Science+Business Media New York 2015 Abstract Polymorphisms in the dopamine D4 receptor Keywords Dopamine Á DRD4 Á Exon III VNTR Á (DRD4) have previously been shown to associate with a variety Neuroscience Á Sexuality of human behavioral phenotypes, including ADHD pathology, alcohol and tobacco craving, financial risk-taking in males, and broader personality traits such as novelty seeking. Recent research has linked the presence of a 7-repeat (7R) allele in a 48- Introduction bp variable number of tandem repeats (VNTR) along exon III of DRD4 to age at first sexual intercourse, sexual desire, arousal and One of many distinguishing behavioral characteristics of our function, and infidelity and promiscuity. We hypothesized that species, human sexuality is evolutionarily novel in a variety of carriers of longer DRD4 alleles may report interest in a wider ways: we exhibit concealed ovulation and menopause, and variety of sexual behaviors and experiences than noncarriers. engage in a wider variety of sexual acts than nonhuman primates Participants completed a 37-item questionnaire measuring sexual (Bancroft, 2009;Dixson,2013), among others. Recent research interests as well as Cloninger’s Temperament and Character has suggested that some variability in human sexuality can be Inventory,andweregenotypedforthe48-bpVNTRonexonIII explained by genetic polymorphisms affecting neuromodula- of DRD4. -

GENETICS the Future of Medicine
GENETICSThe Future of Medicine The Human Genome DNA contains instructions for everything our cells do, from concep- Project tion until death. Studying the human genome—all the DNA in our cells—allows us to explore fundamental details about ourselves. The Exploring Our Human Genome Project, the international quest to understand the Molecular Selves genomes of humans and other organisms, will shed light on a wide range of basic questions, like how many genes we have, how cells work, how living things evolved, how single cells develop into complex creatures, and what exactly happens when we become ill. Besides answering innumerable questions about our molecular selves, a deep- er understanding of the fundamental mechanisms of life promises to lead to an era of molecular medicine, with precise new ways to pre- vent, diagnose and treat disease. The Human Genome Project (HGP) began in the United States in 1990, when the National Institutes of Health and the Department of Energy joined forces with international partners to decipher the massive amount of information contained in our genomes. The HGP began with a set of ambitious goals but has exceeded nearly all of its targets. Frequently ahead of schedule, HGP scientists have produced an increasingly detailed series of maps that help geneticists navi- gate through human DNA. They have mapped and sequenced the genomes of important experimental organisms. They completed a working draft covering 90 percent of the genome in 2000, and by 2003, they will finish the sequence with an accuracy greater than 99.99 percent—fewer than one mistake every 10,000 letters. -

The Enigmatic HOX Genes: Can We Crack Their Code?
cancers Review The Enigmatic HOX Genes: Can We Crack Their Code? Zhifei Luo, Suhn K. Rhie and Peggy J. Farnham * Department of Biochemistry and Molecular Medicine and the Norris Comprehensive Cancer Center, Keck School of Medicine, University of Southern California, Los Angeles, CA 90089, USA; [email protected] (Z.L.); [email protected] (S.K.R.) * Correspondence: [email protected]; Tel.: +323-442-8015 Received: 2 February 2019; Accepted: 1 March 2019; Published: 7 March 2019 Abstract: Homeobox genes (HOX) are a large family of transcription factors that direct the formation of many body structures during early embryonic development. There are 39 genes in the subgroup of homeobox genes that constitute the human HOX gene family. Correct embryonic development of flies and vertebrates is, in part, mediated by the unique and highly regulated expression pattern of the HOX genes. Disruptions in these fine-tuned regulatory mechanisms can lead to developmental problems and to human diseases such as cancer. Unfortunately, the molecular mechanisms of action of the HOX family of transcription factors are severely under-studied, likely due to idiosyncratic details of their structure, expression, and function. We suggest that a concerted and collaborative effort to identify interacting protein partners, produce genome-wide binding profiles, and develop HOX network inhibitors in a variety of human cell types will lead to a deeper understanding of human development and disease. Within, we review the technological challenges and possible approaches needed to achieve this goal. Keywords: HOX; ChIP-seq; cancer biomarkers; targeted therapy 1. Introduction The homeobox gene family is the second largest family of transcription factors encoded in the human genome and consists of an estimated 257 genes [1], each of which contains a 183-nucleotide sequence that encodes a 61-amino acid homeodomain that forms a helix-turn-helix structure.