Chapter 19: Thermochemistry II: Entropy and Free Energy Introduction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump Scott M
Purdue University Purdue e-Pubs CTRC Research Publications Cooling Technologies Research Center 2012 Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump Scott M. Flueckiger Purdue University Fabien Volle Laboratoire des Sciences des Procédés et des Matériaux S V. Garimella Purdue University, [email protected] Rajiv K. Mongia Intel Corporation Follow this and additional works at: http://docs.lib.purdue.edu/coolingpubs Flueckiger, Scott M.; Volle, Fabien; Garimella, S V.; and Mongia, Rajiv K., "Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump" (2012). CTRC Research Publications. Paper 182. http://dx.doi.org/http://dx.doi.org/10.1016/j.enconman.2012.04.015 This document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] for additional information. Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump* Scott M. Flueckiger1, Fabien Volle2, Suresh V. Garimella1**, Rajiv K. Mongia3 1 Cooling Technologies Research Center, an NSF I/UCRC School of Mechanical Engineering and Birck Nanotechnology Center 585 Purdue Mall, Purdue University West Lafayette, Indiana 47907-2088 USA 2 Laboratoire des Sciences des Procédés et des Matériaux (LSPM, UPR 3407 CNRS), Université Paris XIII, 99 avenue J. B. Clément, 93430 Villetaneuse, France 3 Intel Corporation Santa Clara, California 95054 USA * Submitted for publication in Energy Conversion and Management ** Author to who correspondence should be addressed: (765) 494-5621, [email protected] Abstract Representative reversible endothermic chemical reactions (paraldehyde depolymerization and 2-proponal dehydrogenation) are theoretically assessed for their use in a chemical heat pump design for compact thermal management applications. -

Chemical Reactions Involve Energy Changes
Page 1 of 6 KEY CONCEPT Chemical reactions involve energy changes. BEFORE, you learned NOW, you will learn • Bonds are broken and made • About the energy in chemical during chemical reactions bonds between atoms • Mass is conserved in all • Why some chemical reactions chemical reactions release energy • Chemical reactions are • Why some chemical reactions represented by balanced absorb energy chemical equations VOCABULARY EXPLORE Energy Changes bond energy p. 86 How can you identify a transfer of energy? exothermic reaction p. 87 endothermic reaction p. 87 PROCEDURE MATERIALS photosynthesis p. 90 • graduated cylinder 1 Pour 50 ml of hot tap water into the cup • hot tap water and place the thermometer in the cup. • plastic cup 2 Wait 30 seconds, then record the • thermometer temperature of the water. • stopwatch • plastic spoon 3 Measure 5 tsp of Epsom salts. Add the Epsom salts to the beaker and immedi- • Epsom salts ately record the temperature while stirring the contents of the cup. 4 Continue to record the temperature every 30 seconds for 2 minutes. WHAT DO YOU THINK? • What happened to the temperature after you added the Epsom salts? • What do you think caused this change to occur? Chemical reactions release or absorb energy. COMBINATION NOTES Chemical reactions involve breaking bonds in reactants and forming Use combination notes new bonds in products. Breaking bonds requires energy, and forming to organize information on how chemical reactions bonds releases energy. The energy associated with bonds is called bond absorb or release energy. energy. What happens to this energy during a chemical reaction? Chemists have determined the bond energy for bonds between atoms. -

Lecture 7: "Basics of Star Formation and Stellar Nucleosynthesis" Outline
Lecture 7: "Basics of Star Formation and Stellar Nucleosynthesis" Outline 1. Formation of elements in stars 2. Injection of new elements into ISM 3. Phases of star-formation 4. Evolution of stars Mark Whittle University of Virginia Life Cycle of Matter in Milky Way Molecular clouds New clouds with gravitationally collapse heavier composition to form stellar clusters of stars are formed Molecular cloud Stars synthesize Most massive stars evolve He, C, Si, Fe via quickly and die as supernovae – nucleosynthesis heavier elements are injected in space Solar abundances • Observation of atomic absorption lines in the solar spectrum • For some (heavy) elements meteoritic data are used Solar abundance pattern: • Regularities reflect nuclear properties • Several different processes • Mixture of material from many, many stars 5 SolarNucleosynthesis abundances: key facts • Solar• Decreaseabundance in abundance pattern: with atomic number: - Large negative anomaly at Be, B, Li • Regularities reflect nuclear properties - Moderate positive anomaly around Fe • Several different processes 6 - Sawtooth pattern from odd-even effect • Mixture of material from many, many stars Origin of elements • The Big Bang: H, D, 3,4He, Li • All other nuclei were synthesized in stars • Stellar nucleosynthesis ⇔ 3 key processes: - Nuclear fusion: PP cycles, CNO bi-cycle, He burning, C burning, O burning, Si burning ⇒ till 40Ca - Photodisintegration rearrangement: Intense gamma-ray radiation drives nuclear rearrangement ⇒ 56Fe - Most nuclei heavier than 56Fe are due to neutron -

Endothermic and Exothermic Reactions
Name: ________________________ Date: ____________ Period: ____________ Endothermic and Exothermic Reactions Read the following and take notes in the margins. Respond to questions 1-3 at the end. Let's see what Sam and Julie are up to in the chemistry lab. Excited but a bit confused, Sam and Julie run to their chemistry teacher. Sam asks, “Teacher, why did my flask turn cold after adding the salt to water, while Julie’s flask turned hot?” The teacher replies: “That’s because you were given two different salts. One of your salts generated an endothermic reaction with water, while the other salt generated an exothermic reaction with water. Let me first reveal the identity of your salts: Salt A is ammonium nitrate and Salt B is calcium chloride." Now, Sam and Julie are curious about the difference between an endothermic and an exothermic reaction. Consider the reaction mixture—salt plus water—as the system and the flask as the surrounding. In Sam’s case, when ammonium nitrate was dissolved in water, the system absorbed heat from the surrounding, the flask, and thus the flask felt cold. This is an example of an endothermic reaction. In Julie’s case, when calcium chloride was dissolved in water, the system released heat into the surroundings, the flask, and thus the flask felt hot. This is an example of an exothermic reaction. The reaction going on in Sam’s flask can be represented as: NH4NO3 (s) + heat ---> NH4+ (aq) + NO3- (aq) You can see, heat is absorbed during the above reaction, lowering the temperature of the reaction mixture, and thus the reaction flask feels cold. -

Non-Electric Applications of Fusion
Non-Electric Applications of Fusion Final Report to FESAC, July 31, 2003 Executive Summary This report examines the possibility of non-electric applications of fusion. In particular, FESAC was asked to consider “whether the Fusion Energy Sciences program should broaden its scope and activities to include non-electric applications of intermediate-term fusion devices.” During this process, FESAC was asked to consider the following questions: • What are the most promising opportunities for using intermediate-term fusion devices to contribute to the Department of Energy missions beyond the production of electricity? • What steps should the program take to incorporate these opportunities into plans for fusion research? • Are there any possible negative impacts to pursuing these opportunities and are there ways to mitigate these possible impacts? The panel adopted the following three criteria to evaluate all of the non-electric applications considered: 1. Will the application be viewed as necessary to solve a "national problem" or will the application be viewed as a solution by the funding entity? 2. What are the technical requirements on fusion imposed by this application with respect to the present state of fusion and the technical requirements imposed by electricity production? What R&D is required to meet these requirements and is it "on the path" to electricity production? 3. What is the competition for this application, and what is the likelihood that fusion can beat it? It is the opinion of this panel that the most promising opportunities for non-electric applications of fusion fall into four categories: 1. Near-Term Applications 2. Transmutation 3. Hydrogen Production 4. -

Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions
CHEM 1B: GENERAL CHEMISTRY Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions Instructor: Dr. Orlando E. Raola 20-1 Santa Rosa Junior College Chapter 20 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20-2 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20.1 The Second Law of Thermodynamics: Predicting Spontaneous Change 20.2 Calculating Entropy Change of a Reaction 20.3 Entropy, Free Energy, and Work 20.4 Free Energy, Equilibrium, and Reaction Direction 20-3 Spontaneous Change A spontaneous change is one that occurs without a continuous input of energy from outside the system. All chemical processes require energy (activation energy) to take place, but once a spontaneous process has begun, no further input of energy is needed. A nonspontaneous change occurs only if the surroundings continuously supply energy to the system. If a change is spontaneous in one direction, it will be nonspontaneous in the reverse direction. 20-4 The First Law of Thermodynamics Does Not Predict Spontaneous Change Energy is conserved. It is neither created nor destroyed, but is transferred in the form of heat and/or work. DE = q + w The total energy of the universe is constant: DEsys = -DEsurr or DEsys + DEsurr = DEuniv = 0 The law of conservation of energy applies to all changes, and does not allow us to predict the direction of a spontaneous change. 20-5 DH Does Not Predict Spontaneous Change A spontaneous change may be exothermic or endothermic. Spontaneous exothermic processes include: • freezing and condensation at low temperatures, • combustion reactions, • oxidation of iron and other metals. -

Chemical Engineering Vocabulary
Chemical Engineering Vocabulary Maximilian Lackner Download free books at MAXIMILIAN LACKNER CHEMICAL ENGINEERING VOCABULARY Download free eBooks at bookboon.com 2 Chemical Engineering Vocabulary 1st edition © 2016 Maximilian Lackner & bookboon.com ISBN 978-87-403-1427-4 Download free eBooks at bookboon.com 3 CHEMICAL ENGINEERING VOCABULARY a.u. (sci.) Acronym/Abbreviation referral: see arbitrary units A/P (econ.) Acronym/Abbreviation referral: see accounts payable A/R (econ.) Acronym/Abbreviation referral: see accounts receivable abrasive (eng.) Calcium carbonate can be used as abrasive, for example as “polishing agent” in toothpaste. absorbance (chem.) In contrast to absorption, the absorbance A is directly proportional to the concentration of the absorbing species. A is calculated as ln (l0/l) with l0 being the initial and l the transmitted light intensity, respectively. absorption (chem.) The absorption of light is often called attenuation and must not be mixed up with adsorption, an effect at the surface of a solid or liquid. Absorption of liquids and gases means that they diffuse into a liquid or solid. abstract (sci.) An abstract is a summary of a scientific piece of work. AC (eng.) Acronym/Abbreviation referral: see alternating current academic (sci.) The Royal Society, which was founded in 1660, was the first academic society. acceleration (eng.) In SI units, acceleration is measured in meters/second Download free eBooks at bookboon.com 4 CHEMICAL ENGINEERING VOCABULARY accompanying element (chem.) After precipitation, the thallium had to be separated from the accompanying elements. TI (atomic number 81) is highly toxic and can be found in rat poisons and insecticides. accounting (econ.) Working in accounting requires paying attention to details. -
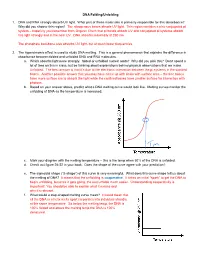
DNA Folding/Unfolding 1. DNA and RNA Strongly Absorb UV Light. What Part of These Molecules Is Primarily Responsible for This A
DNA Folding/Unfolding 1. DNA and RNA strongly absorb UV light. What part of these molecules is primarily responsible for this absorbance? Why did you choose this region? The nitrogenous bases absorb UV light. This region contains a nice conjugated pi system – hopefully you remember from Organic Chem that pi bonds absorb UV and conjugated pi systems absorb this light strongly and in the near UV. DNA absorbs maximally at 260 nm. The phosphate backbone also absorbs UV light, but at much lower frequencies. 2. The hyperchromic effect is used to study DNA melting. This is a general phenomenon that explains the difference in absorbance between folded and unfolded DNA and RNA molecules. a. Which absorbs light more strongly: folded or unfolded nucleic acids? Why did you pick this? Don’t spend a lot of time on this in class, but be thinking about explanations behind physical observations that we make. Unfolded. The best answer is that it’s due to the electronic interaction between the pi systems in the stacked bases. Another possible answer that you may have come up with deals with surface area – the free bases have more surface are to absorb the light while the confined bases have smaller surface for interaction with photons. b. Based on your answer above, predict what a DNA melting curve would look like. Melting curves monitor the unfolding of DNA as the temperature is increased. c. Mark your diagram with the melting temperature – this is the temp when 50% of the DNA is unfolded. d. Check out figure 24-22 in your book. -

Endothermic & Exothermic Reactions
Endothermic & Exothermic Reactions Written by Chris Papadopoulos This lesson focuses on the use of technology to collect, graph and analyze data from an exothermic and an endothermic reaction. Hypothesis Chemical reactions are systems that exchange heat/energy with their surroundings. Primary Learning Outcomes At the end of this lesson, students will be able to: • Be familiarized with data collection using the Vernier LabPro and TI-84 calculator • Collect, graph, display and make inferences from data • Calculate the temperature change (∆t) of a reaction • Determine whether a reaction is exothermic or endothermic based on ∆t Assessed GPS Characteristics of Science: Habits of Mind: SCSh2. Students will use standard safety practices for all classroom laboratory and field investigations. a. Follow correct procedures for use of scientific apparatus. b. Demonstrate appropriate techniques in all laboratory situations. c. Follow correct protocol for identifying and reporting safety problems and violations. SCSh3. Students will identify and investigate problems scientifically c. Collect, organize, and record appropriate data d. Graphically compare and analyze data points and/or summary statistics e. Develop reasonable conclusions based on data collected SCSh4. Students will use tools and instruments for observing, measuring, and manipulating scientific equipment and materials. b. Use technology to produce tables and graphs SCSh5. Students will demonstrate the computation and estimation skills necessary for analyzing data and developing reasonable scientific explanations. e. Solve scientific problems by substituting quantitative values, using dimensional analysis and/or simple algebraic formulas as appropriate. Content: SC2. Students will relate how the Law of Conservation of Matter is used to determine chemical composition in compounds and chemical reactions. -

Information Sheet
Information Sheet Heat, enthalpy and entropy Energy considerations play a central role in virtually all processes in science and technology: Does one need to add energy to a chemical reaction so that it takes place, or does it run spontaneously and with the release of energy? How much energy is released in an organic cell as a product of metabolism? How does an engine need to be designed so that the energy of the fuel is converted into mechanical energy with optimum effect? Different terms are used for “energy” depending on the special field. In chemistry, for example, one speaks of the “enthalpy” or the “free enthalpy” of a chemical reaction, while in thermodynamics “internal energy” and “heat” are the key parameters. How are all these terms related and what do they state? Heat and enthalpy are energy quantities Heat Heat (Q) is an amount of energy. It is equivalent to the energy that is needed to raise a system from a lower temperature to a higher temperature. The greater the temperature difference (T) is to be, the more heat has to be input – the two quantities are directly proportional to one another. Exactly how much energy needs to be expended depends on the material. This material property is called “heat capacity” C. Referred to the mass (m) of the material, one speaks of the “specific heat capacity” (also shortened to “specific heat” which can be misunderstood): c = C/m. The heat energy to be expended (“heat” for short) can thus be calculated via the formula: Q = C T = c m T Water, for example, has a specific heat capacity cH2O = 4.183 kJ/ (kg K) (= 1 kcal/ (kg K) ), i.e. -

Chemical Reaction Lesson Plan
ENDO AND EXOTHERMIC REACTIONS Description Overview: A chemical reaction is a process that leads to the chemical transformation of one set of chemical substances to another. Chemical reactions are an integral part of technology, of culture, and indeed of life itself. Burning fuels, smelting iron, making glass and pottery, brewing beer, and making wine and cheese are among many examples of activities incorporating chemical reactions that have been known and used for thousands of years. Chemical reactions abound in the geology of Earth, in the atmosphere and oceans, and in a vast array of complicated processes that occur in all living systems. Chemical reactions must be distinguished from physical changes. Physical changes include changes of state, such as ice melting to water and water evaporating to vapor. If a physical change occurs, the physical properties of a substance will change, but its chemical identity will remain the same. No matter what its physical state, water (H2O) is the same compound, with each molecule composed of two atoms of hydrogen and one atom of oxygen. However, if water, as ice, liquid, or vapor, encounters sodium metal (Na), the atoms will be redistributed to give the new substances molecular hydrogen (H2) and sodium hydroxide (NaOH). By this, we know that a chemical change or reaction has occurred. Exothermic Reactions. Exothermic reactions are reactions or processes that release energy, usually in the form of heat or light. In an exothermic reaction, energy is released because the total energy of the products is less than the total energy of the reactants. An endothermic reaction is any chemical reaction that absorbs heat from its environment. -

Answers to for Review Questions from the Textbook
CHAPTER SIX THERMOCHEMISTRY For Review 1. Potential energy: energy due to position or composition Kinetic energy: energy due to motion of an object Path-dependent function: a property that depends on how the system gets from the initial state to the final state; a property that is path-dependent State function: a property that is independent of the pathway System: that part of the universe on which attention is to be focused Surroundings: everything in the universe surrounding a thermodynamic system 2. Plot a represents an exothermic reaction. In an exothermic process, the bonds in the product molecules are stronger (on average) than those in the reactant molecules. The net result is that the quantity of energy ∆(PE) is transferred to the surroundings as heat when reactants are converted to products. For an endothermic process, energy flows into the system from the surroundings as heat to increase the potential energy of the system. In an endothermic process, the products have higher potential energy (weaker bonds on average) than the reactants. 3. First law of thermodynamics: the energy of universe is constant. A system can change its internal energy by flow of work, heat, or both (∆E = q + w). Whenever a property is added to the system from the surroundings, the sign is positive; whenever a property is added to the surroundings by the system, the sign is negative. 4. As a gas expands, the system does work on the surroundings so w is negative. When a gas contracts, the surroundings do work on the system so w is positive. H2O(l) → H2O(g); To boil water, heat must be added so q is positive.