Entropy Pair Functional Theory: Direct Entropy Evaluation Spanning Phase Transitions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2D Potts Model Correlation Lengths
KOMA Revised version May D Potts Mo del Correlation Lengths Numerical Evidence for = at o d t Wolfhard Janke and Stefan Kappler Institut f ur Physik Johannes GutenbergUniversitat Mainz Staudinger Weg Mainz Germany Abstract We have studied spinspin correlation functions in the ordered phase of the twodimensional q state Potts mo del with q and at the rstorder transition p oint Through extensive Monte t Carlo simulations we obtain strong numerical evidence that the cor relation length in the ordered phase agrees with the exactly known and recently numerically conrmed correlation length in the disordered phase As a byproduct we nd the energy moments o t t d in the ordered phase at in very go o d agreement with a recent large t q expansion PACS numbers q Hk Cn Ha hep-lat/9509056 16 Sep 1995 Introduction Firstorder phase transitions have b een the sub ject of increasing interest in recent years They play an imp ortant role in many elds of physics as is witnessed by such diverse phenomena as ordinary melting the quark decon nement transition or various stages in the evolution of the early universe Even though there exists already a vast literature on this sub ject many prop erties of rstorder phase transitions still remain to b e investigated in detail Examples are nitesize scaling FSS the shap e of energy or magnetization distributions partition function zeros etc which are all closely interrelated An imp ortant approach to attack these problems are computer simulations Here the available system sizes are necessarily -

Full and Unbiased Solution of the Dyson-Schwinger Equation in the Functional Integro-Differential Representation
PHYSICAL REVIEW B 98, 195104 (2018) Full and unbiased solution of the Dyson-Schwinger equation in the functional integro-differential representation Tobias Pfeffer and Lode Pollet Department of Physics, Arnold Sommerfeld Center for Theoretical Physics, University of Munich, Theresienstrasse 37, 80333 Munich, Germany (Received 17 March 2018; revised manuscript received 11 October 2018; published 2 November 2018) We provide a full and unbiased solution to the Dyson-Schwinger equation illustrated for φ4 theory in 2D. It is based on an exact treatment of the functional derivative ∂/∂G of the four-point vertex function with respect to the two-point correlation function G within the framework of the homotopy analysis method (HAM) and the Monte Carlo sampling of rooted tree diagrams. The resulting series solution in deformations can be considered as an asymptotic series around G = 0 in a HAM control parameter c0G, or even a convergent one up to the phase transition point if shifts in G can be performed (such as by summing up all ladder diagrams). These considerations are equally applicable to fermionic quantum field theories and offer a fresh approach to solving functional integro-differential equations beyond any truncation scheme. DOI: 10.1103/PhysRevB.98.195104 I. INTRODUCTION was not solved and differences with the full, exact answer could be seen when the correlation length increases. Despite decades of research there continues to be a need In this work, we solve the full DSE by writing them as a for developing novel methods for strongly correlated systems. closed set of integro-differential equations. Within the HAM The standard Monte Carlo approaches [1–5] are convergent theory there exists a semianalytic way to treat the functional but suffer from a prohibitive sign problem, scaling exponen- derivatives without resorting to an infinite expansion of the tially in the system volume [6]. -

Feas: Heat Capacity, Enthalpy Increments, Other Thermodynamic Properties from 5 to 1350 K, and Magnetic Transition A
M-2341 J. Chem. Thermodynumics 1989,21, 363-373 FeAs: Heat capacity, enthalpy increments, other thermodynamic properties from 5 to 1350 K, and magnetic transition a DOMINGO GONZALEZ-ALVAREZ, Fact&ad de Ciencias de la Universidad de Zaragoza, Departamento de Fisica Fundamental, 5009 Zaragoza, Spain FREDRIK GR0NVOLD, Chemical Institute, University of Oslo, Blindern, Oslo 3, Norway BENGT FALK,b EDGAR F. WESTRUM, JR., Department of Chemistry, University of Michigan, Ann Arbor, MI 48109, U.S.A. R. BLACHNIK,’ and G. KUDERMANNd Technical University, Siegen, Federal Republic of Germany (Received 2 January 1989) The heat capacity of iron monoarsenide has been determined by adiabatic calorimetry from 5 to 1030 K and bv drou calorimetrv relative to 298.15 K over the range 875 to 1350 K. A small h-type transition is observed at ?;, = (70.95 kO.02) K. It is related & the disappearance of a doubly helically ordered magnetic-spin structure on heating. The obviously cooperative entropy increment of transition is only A&JR = 0.021. The higher-temperature heat capacity rises considerably above lattice expectations. Part of the rise is ascribed to low-spin electron redistribution in iron, while the further excess above 800 K presumably arises from a beginning low- to high-spin transition, possibly connected with interstitial defect formation in the MnP- type structure. FeAs melts at about 1325 K with A,,,Hk=6180R.K. Thermodynamic functions have been evaluated and the values of C,,,(T), S:(T), H:(T), and @i(T), are 6.057R, 7.513R,1177R.K, and 3.567Rat 298.15 K, and 8.75R,16.03R, 6287R.K, and 9.74512at 1000 K. -

Notes on Statistical Field Theory
Lecture Notes on Statistical Field Theory Kevin Zhou [email protected] These notes cover statistical field theory and the renormalization group. The primary sources were: • Kardar, Statistical Physics of Fields. A concise and logically tight presentation of the subject, with good problems. Possibly a bit too terse unless paired with the 8.334 video lectures. • David Tong's Statistical Field Theory lecture notes. A readable, easygoing introduction covering the core material of Kardar's book, written to seamlessly pair with a standard course in quantum field theory. • Goldenfeld, Lectures on Phase Transitions and the Renormalization Group. Covers similar material to Kardar's book with a conversational tone, focusing on the conceptual basis for phase transitions and motivation for the renormalization group. The notes are structured around the MIT course based on Kardar's textbook, and were revised to include material from Part III Statistical Field Theory as lectured in 2017. Sections containing this additional material are marked with stars. The most recent version is here; please report any errors found to [email protected]. 2 Contents Contents 1 Introduction 3 1.1 Phonons...........................................3 1.2 Phase Transitions......................................6 1.3 Critical Behavior......................................8 2 Landau Theory 12 2.1 Landau{Ginzburg Hamiltonian.............................. 12 2.2 Mean Field Theory..................................... 13 2.3 Symmetry Breaking.................................... 16 3 Fluctuations 19 3.1 Scattering and Fluctuations................................ 19 3.2 Position Space Fluctuations................................ 20 3.3 Saddle Point Fluctuations................................. 23 3.4 ∗ Path Integral Methods.................................. 24 4 The Scaling Hypothesis 29 4.1 The Homogeneity Assumption............................... 29 4.2 Correlation Lengths.................................... 30 4.3 Renormalization Group (Conceptual).......................... -

High Temperature Enthalpies of the Lead Halides
HIGH TEMPERATURE ENTHALPIES OF THE LEAD HALIDES: ENTHALPIES AND ENTROPIES OF FUSION APPROVED: Graduate Committee: Major Professor Committee Member Min fessor Committee Member X Committee Member UJ. Committee Member Committee Member Chairman of the Department of Chemistry Dean or the Graduate School HIGH TEMPERATURE ENTHALPIES OF THE LEAD HALIDES: ENTHALPIES AND ENTROPIES OF FUSION DISSERTATION Presented to the Graduate Council of the North Texas State University in Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY By Clarence W, Linsey, B. S., M. S, Denton, Texas June, 1970 ACKNOWLEDGEMENT This dissertation is based on research conducted at the Oak Ridge National Laboratory, Oak Ridge, Tennessee, which is operated by the Uniofl Carbide Corporation for the U. S. Atomic Energy Commission. The author is also indebted to the Oak Ridge Associated Universities for the Oak Ridge Graduate Fellowship which he held during the laboratory phase. 11 TABLE OF CONTENTS Page LIST OF TABLES iv LIST OF ILLUSTRATIONS v Chapter I. INTRODUCTION . 1 Lead Fluoride Lead Chloride Lead Bromide Lead Iodide II. EXPERIMENTAL 11 Reagents Fusion-Filtration Method Encapsulation of Samples Equipment and Procedure Bunsen Ice Calorimeter Computer Programs Shomate Method Treatment of Data Near the Melting Point Solution Calorimeter Heat of Solution Calculations III. RESULTS AND DISCUSSION 41 Lead Fluoride Variation in Heat Contents of the Nichrome V Capsules Lead Chloride Lead Bromide Lead Iodide Summary BIBLIOGRAPHY ...... 98 in LIST OF TABLES Table Pa8e I. High-Temperature Enthalpy Data for Cubic PbE2 Encapsulated in Gold 42 II. High-Temperature Enthalpy Data for Cubic PbF2 Encapsulated in Molybdenum 48 III. -

10 Basic Aspects of CFT
10 Basic aspects of CFT An important break-through occurred in 1984 when Belavin, Polyakov and Zamolodchikov [BPZ84] applied ideas of conformal invariance to classify the possible types of critical behaviour in two dimensions. These ideas had emerged earlier in string theory and mathematics, and in fact go backto earlier (1970) work of Polyakov [Po70] in which global conformal invariance is used to constrain the form of correlation functions in d-dimensional the- ories. It is however only by imposing local conformal invariance in d =2 that this approach becomes really powerful. In particular, it immediately permitted a full classification of an infinite family of conformally invariant theories (the so-called “minimal models”) having a finite number of funda- mental (“primary”) fields, and the exact computation of the corresponding critical exponents. In the aftermath of these developments, conformal field theory (CFT) became for some years one of the most hectic research fields of theoretical physics, and indeed has remained a very active area up to this date. This chapter focusses on the basic aspects of CFT, with a special emphasis on the ingredients which will allow us to tackle the geometrically defined loop models via the so-called Coulomb Gas (CG) approach. The CG technique will be exposed in the following chapter. The aim is to make the presentation self- contained while remaining rather brief; the reader interested in more details should turn to the comprehensive textbook [DMS87] or the Les Houches volume [LH89]. 10.1 Global conformal invariance A conformal transformation in d dimensions is an invertible mapping x x′ → which multiplies the metric tensor gµν (x) by a space-dependent scale factor: gµ′ ν (x′)=Λ(x)gµν (x). -

THERMODYNAMIC EVALUATION of the Cu-Ti SYSTEM in VIEW of SOLID STATE AMORPHIZATION REACTIONS L
THERMODYNAMIC EVALUATION OF THE Cu-Ti SYSTEM IN VIEW OF SOLID STATE AMORPHIZATION REACTIONS L. Battezzati, M. Baricco, G. Riontino, I. Soletta To cite this version: L. Battezzati, M. Baricco, G. Riontino, I. Soletta. THERMODYNAMIC EVALUATION OF THE Cu- Ti SYSTEM IN VIEW OF SOLID STATE AMORPHIZATION REACTIONS. Journal de Physique Colloques, 1990, 51 (C4), pp.C4-79-C4-85. 10.1051/jphyscol:1990409. jpa-00230769 HAL Id: jpa-00230769 https://hal.archives-ouvertes.fr/jpa-00230769 Submitted on 1 Jan 1990 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ~OLLOQUEDE PHYSIQUE Colloque C4, suppl6ment au 11'14, Tome 51, 15 juillet 1990 THERMODYNAMIC EVALUATION OF THE Cu-Ti SYSTEM IN VIEW OF SOLID STATE AMORPHIZATION REACTIONS L. BATTEZZATI* , M. BARICCO*~ *""" , G. RIONTINO" *"* and I. SOLETTA** * '~ipartimento di Chimica Inorganics, Chimica Fisica e Chimica dei Materiali, Universitd di Torino, Italy ""~stitutoElettrotecnico Nazionale Galileo Perraris, Torino, Italy *.* Dipartimento di Chimica, Universitd di Sassari, Italy *et* INPM, Unita' di Ricerca di Torino, Italy Resumk -La description thermodynamique du sist6me Cu-Ti est reconsideree, car les calculs he la littgrature, bien que donnant une bonne representation du diagramme de phase, ne prevoient pas la possibilit6 de l'amorphisation. -

Introduction to Two-Dimensional Conformal Field Theory
Introduction to two-dimensional conformal field theory Sylvain Ribault CEA Saclay, Institut de Physique Th´eorique [email protected] February 5, 2019 Abstract We introduce conformal field theory in two dimensions, from the basic principles to some of the simplest models. From the representations of the Virasoro algebra on the one hand, and the state-field correspondence on the other hand, we deduce Ward identities and Belavin{Polyakov{Zamolodchikov equations for correlation functions. We then explain the principles of the conformal bootstrap method, and introduce conformal blocks. This allows us to define and solve minimal models and Liouville theory. We also introduce the free boson with its abelian affine Lie algebra. Lecture notes for the \Young Researchers Integrability School", Wien 2019, based on the earlier lecture notes [1]. Estimated length: 7 lectures of 45 minutes each. Material that may be skipped in the lectures is in green boxes. Exercises are in green boxes when their statements are not part of the lectures' text; this does not make them less interesting as exercises. 1 Contents 0 Introduction2 1 The Virasoro algebra and its representations3 1.1 Algebra.....................................3 1.2 Representations.................................4 1.3 Null vectors and degenerate representations.................5 2 Conformal field theory6 2.1 Fields......................................6 2.2 Correlation functions and Ward identities...................8 2.3 Belavin{Polyakov{Zamolodchikov equations................. 10 2.4 Free boson.................................... 10 3 Conformal bootstrap 12 3.1 Single-valuedness................................ 12 3.2 Operator product expansion and crossing symmetry............. 13 3.3 Degenerate fields and the fusion product................... 16 4 Minimal models 17 4.1 Diagonal minimal models........................... -
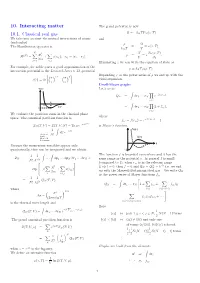
10. Interacting Matter 10.1. Classical Real
10. Interacting matter The grand potential is now Ω= k TVω(z,T ) 10.1. Classical real gas − B We take into account the mutual interactions of atoms and (molecules) p Ω The Hamiltonian operator is = = ω(z,T ) kBT −V N p2 N ∂ω(z,T ) H(N) = i + v(r ), r = r r . ρ = = z . 2m ij ij | i − j | V ∂z i=1 i<j X X Eliminating z we can write the equation of state as For example, for noble gases a good approximation of the p = k T ϕ(ρ,T ). interaction potential is the Lennard-Jones 6–12 -potential B Expanding ϕ as the power series of ρ we end up with the σ 12 σ 6 v(r)=4ǫ . virial expansion. r − r Ursell-Mayer graphs V ( r ) Let’s write Q = dr r e−βv(rij ) N 1 · · · N i<j Z Y r 0 s r 6 = dr r (1 + f ), e 1 / r 1 · · · N ij Z i<j We evaluate the partition sums in the classical phase Y where space. The canonical partition function is f = f(r )= e−βv(rij ) 1 ij ij − −βH(N) ZN (T, V )= Z(T,V,N) = Tr N e is Mayer’s function. 1 V ( r ) dΓ e−βH . classical−→ N! limit, Maxwell- Z f ( r ) Boltzman Because the momentum variables appear only r quadratically, they can be integrated and we obtain - 1 1 1 The function f is bounded everywhere and it has the ZN = dp1 dpN dr1 drN same range as the potential v. -

Recursive Graphical Solution of Closed Schwinger-Dyson
Recursive Graphical Solution of Closed Schwinger-Dyson Equations in φ4-Theory – Part1: Generation of Connected and One-Particle Irreducible Feynman Diagrams Axel Pelster and Konstantin Glaum Institut f¨ur Theoretische Physik, Freie Universit¨at Berlin, Arnimallee 14, 14195 Berlin, Germany [email protected],[email protected] (Dated: November 11, 2018) Using functional derivatives with respect to the free correlation function we derive a closed set of Schwinger-Dyson equations in φ4-theory. Its conversion to graphical recursion relations allows us to systematically generate all connected and one-particle irreducible Feynman diagrams for the two- and four-point function together with their weights. PACS numbers: 05.70.Fh,64.60.-i I. INTRODUCTION Quantum and statistical field theory investigate the influence of field fluctuations on the n-point functions. Interactions lead to an infinite hierarchy of Schwinger-Dyson equations for the n-point functions [1–6]. These integral equations can only be closed approximately, for instance, by the well-established the self-consistent method of Kadanoff and Baym [7]. Recently, it has been shown that the Schwinger-Dyson equations of QED can be closed in a certain functional- analytic sense [8]. Using functional derivatives with respect to the free propagators and the interaction [8–14] two closed sets of equations were derived. The first one involves the connected electron and two-point function as well as the connected three-point function, whereas the second one determines the electron and photon self-energy as well as the one-particle irreducible three-point function. Their conversion to graphical recursion relations leads to a systematic graphical generation of all connected and one-particle irreducible Feynman diagrams in QED, respectively. -

Efficient Approach to Compute Melting Properties Fully from Ab Initio With
PHYSICAL REVIEW B 96, 224202 (2017) Efficient approach to compute melting properties fully from ab initio with application to Cu Li-Fang Zhu,* Blazej Grabowski, and Jörg Neugebauer Max-Planck-Institut für Eisenforschung GmbH, D-40237 Düsseldorf, Germany (Received 3 August 2017; revised manuscript received 16 October 2017; published 13 December 2017) Applying thermodynamic integration within an ab initio-based free-energy approach is a state-of-the-art method to calculate melting points of materials. However, the high computational cost and the reliance on a good reference system for calculating the liquid free energy have so far hindered a general application. To overcome these challenges, we propose the two-optimized references thermodynamic integration using Langevin dynamics (TOR-TILD) method in this work by extending the two-stage upsampled thermodynamic integration using Langevin dynamics (TU-TILD) method, which has been originally developed to obtain anharmonic free energies of solids, to the calculation of liquid free energies. The core idea of TOR-TILD is to fit two empirical potentials to the energies from density functional theory based molecular dynamics runs for the solid and the liquid phase and to use these potentials as reference systems for thermodynamic integration. Because the empirical potentials closely reproduce the ab initio system in the relevant part of the phase space the convergence of the thermodynamic integration is very rapid. Therefore, the proposed approach improves significantly the computational efficiency while preserving the required accuracy. As a test case, we apply TOR-TILD to fcc Cu computing not only the melting point but various other melting properties, such as the entropy and enthalpy of fusion and the volume change upon melting. -

Dirty Tricks for Statistical Mechanics
Dirty tricks for statistical mechanics Martin Grant Physics Department, McGill University c MG, August 2004, version 0.91 ° ii Preface These are lecture notes for PHYS 559, Advanced Statistical Mechanics, which I’ve taught at McGill for many years. I’m intending to tidy this up into a book, or rather the first half of a book. This half is on equilibrium, the second half would be on dynamics. These were handwritten notes which were heroically typed by Ryan Porter over the summer of 2004, and may someday be properly proof-read by me. Even better, maybe someday I will revise the reasoning in some of the sections. Some of it can be argued better, but it was too much trouble to rewrite the handwritten notes. I am also trying to come up with a good book title, amongst other things. The two titles I am thinking of are “Dirty tricks for statistical mechanics”, and “Valhalla, we are coming!”. Clearly, more thinking is necessary, and suggestions are welcome. While these lecture notes have gotten longer and longer until they are al- most self-sufficient, it is always nice to have real books to look over. My favorite modern text is “Lectures on Phase Transitions and the Renormalisation Group”, by Nigel Goldenfeld (Addison-Wesley, Reading Mass., 1992). This is referred to several times in the notes. Other nice texts are “Statistical Physics”, by L. D. Landau and E. M. Lifshitz (Addison-Wesley, Reading Mass., 1970) par- ticularly Chaps. 1, 12, and 14; “Statistical Mechanics”, by S.-K. Ma (World Science, Phila., 1986) particularly Chaps.