Autosomal Recessive, Early-Onset Parkinson's Disease
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Protein PARK7 (DJ-1)
Catalogue # Aliquot Size P219-31H-20 20 µg P219-31H-50 50 µg PARK7 (DJ-1) Protein Recombinant protein expressed in E.coli cells Catalog # P219-31H Lot # F495 -3 Product Description Purity Recombinant human PARK7 (DJ-1) (19-end) was expressed in E. coli cells using an N-terminal His tag. The gene accession number is NM_007262 . The purity of PARK7 (DJ-1) was Gene Aliases determined to be >85% by densitometry. PARK7; DJ-1; DJ1 Approx. MW 22 kDa . Formulation Recombinant protein stored in 50mM sodium phosphate, pH 7.0, 300mM NaCl, 150mM imidazole, 0.1mM PMSF, 0.25mM DTT, 25% glycerol. Storage and Stability o Store product at –70 C. For optimal storage, aliquot target into smaller quantities after centrifugation and store at recommended temperature. For most favorable performance, avoid repeated handling and multiple freeze/thaw cycles. Scientific Background PARK7 or parkinson protein 7 belongs to the peptidase C56 family of proteins which acts as a positive regulator of androgen receptor-dependent transcription. PARK7 also functions as a redox-sensitive chaperone, as a sensor for oxidative stress, and it apparently protects neurons PARK7 (DJ-1) Protein against oxidative stress and cell death. PARK7 mutations Recombinant protein expressed in E. coli cells that impair transcriptional co-activator function can render dopaminergic neurons vulnerable to apoptosis Catalog Number P219-31H and may contribute to the pathogenesis of Parkinson Specific Lot Number F495-3 disease (1). PARK7 is an atypical peroxiredoxin-like Purity >85% peroxidase that scavenges hydrogen peroxide through Concentration 0.2µg/ µl Stability 1yr At –70 oC from date of shipment oxidation of cys106 (2). -

(PARK7), a Novel Gene for Autosomal Recessive, Early Onset
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Erasmus University Digital Repository Neurol Sci (2003) 24:159–160 DOI 10.1007/s10072-003-0108-0 DJ-1 (PARK7), a novel gene for Fine mapping studies and a positional cloning strategy autosomal recessive, early onset led us to the identification of homozygous mutations in the DJ-1 gene showing complete cosegregation with the disease parkinsonism haplotype and absence from large numbers of control chro- 1,2 1 3 mosomes: a ~14-kb deletion removing a large part of the DJ- V. Bonifati (౧) • P. Rizzu • F. Squitieri 4 5 6 1 coding region in the Dutch family and a missense mutation E. Krieger • N. Vanacore • J.C. van Swieten 7 8 1 2 (Leucine166Proline, L166P) in the Italian family (Fig. 1) [4]. A. Brice • C.M. van Duijn • B. Oostra • G. Meco 1 The expression of the DJ-1 gene is abolished by the P. Heutink homozygous deletion in the patients of the Dutch family, 1 Department of Clinical Genetics, Erasmus Medical Center, P.O. Box 1738, DR Rotterdam, The Netherlands indicating that the loss of DJ-1 function is pathogenic. The 2 Dipartimento di Scienze Neurologiche, Università “La Sapienza”, L166P mutation is also likely to severely affect the function Rome, Italy of DJ-1 because: (1) it replaces a highly conserved residue in 3 Neurogenetics Unit, IRCCS Neuromed, Pozzilli, Italy the DJ-1 protein, (2) it destabilizes the carboxy-terminal a- 4 Center for Molecular and Biomolecular Informatics, University helix of the DJ-1 protein as predicted by structural models, Medical Center Nijmegen, The Netherlands and (3) it dramatically changes the subcellular localization of 5 Department of Epidemiology and Biostatistics, National Institute the DJ-1 protein in transfection experiments [4]. -

DJ-1 Binds to Mitochondrial Complex I and Maintains Its Activity
Title DJ-1 binds to mitochondrial complex I and maintains its activity Hayashi, Takuya; Ishimori, Chikako; Takahashi-Niki, Kazuko; Taira, Takahiro; Kim, Yun-chul; Maita, Hiroshi; Maita, Author(s) Chinatsu; Ariga, Hiroyoshi; Iguchi-Ariga, Sanae M. M. Biochemical and Biophysical Research Communications, 390(3), 667-672 Citation https://doi.org/10.1016/j.bbrc.2009.10.025 Issue Date 2009-12-18 Doc URL http://hdl.handle.net/2115/42484 Type article (author version) File Information BBRC390-3_667-672.pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP *Manuscript DJ-1 binds to mitochondrial complex I and maintains its activity Takuya Hayashia, Chikako Ishimoria, Kazuko Takahashi-Nikic, Takahiro Tairab, Yun-chul Kimc, Hiroshi Maitac, Chinatsu Maitaa, Hiroyoshi Arigac, *, Sanae M.M. Iguchi-Arigaa aGraduate School of Agriculture, Hokkaido University, Sapporo 060-8589, Japan bInterdisciplinary Graduate School of Medicine and Engineering, Yamanashi University, Chuoh, 409-3898, Japan cGraduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan *Correspondence author. Address: Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan. Fax: +81 11 706 3745. E-mail address: [email protected] (H. Ariga) 1 ABSTRACT Parkinson’s disease (PD) is caused by neuronal cell death, and oxidative stress and mitochondrial dysfunction are thought to be responsible for onset of PD. DJ-1, a causative gene product of a familial form of Parkinson’s disease, PARK7, plays roles in transcriptional regulation and anti-oxidative stress. The possible mitochondrial function of DJ-1 has been proposed, but its exact function remains unclear. In this study, we found that DJ-1 directly bound to NDUFA4 and ND1, nuclear and mitochondrial DNA-encoding subunits of mitochondrial complex I, respectively, and was co-localized with complex I and that complex I activity was reduced in DJ-1-knockdown NIH3T3 and HEK293cells. -

Human Park7/DJ-1 Antibody Catalog Number: ATGA0292
Human Park7/DJ-1 antibody Catalog Number: ATGA0292 PRODUCT INPORMATION Catalog number ATGA0292 Clone No. AT1E12 Product type Monoclonal Antibody UnitProt No. Q99497 NCBI Accession No. NP_009193 Alternative Names Parkinson disease protein 7, PARK7, DJ-1 (PARK7), Oncogene DJ1, Parkinson disease protein 7, Protein DJ-1, Parkinson disease protein 7 CAP1, DJ1, DJ1 protein, Oncogene DJ1, SP22 PRODUCT SPECIFICATION Antibody Host Mouse Reacts With Human Concentration 1mg/ml (determined by BCA assay) Formulation Liquid in. Phosphate-Buffered Saline (pH 7.4) with 0.02% Sodium Azide, 10% glycerol Immunogen Recombinant human Park7/DJ-1 (1-189aa) purified from E. coli Isotype IgG2b kappa Purification Note By protein-A affinity chromatography Application ELISA,WB,ICC/IF,FACS Usage The antibody has been tested by ELISA, Western blot, ICC/IF and FACS analysis to assure specificity and reactivity. Since application varies, however, each investigation should be titrated by the reagent to obtain optimal results. 1 Human Park7/DJ-1 antibody Catalog Number: ATGA0292 Storage Can be stored at +2C to +8C for 1 week. For long term storage, aliquot and store at -20C to -80C. Avoid repeated freezing and thawing cycles. BACKGROUND Description Parkinson disease (autosomal recessive, early onset) 7, also known as PARK7/DJ-1, has been shown to interact with EFCAB6 and protein inhibitor of activated STAT2. Defects in PARK7 are the cause of autosomal recessive early-onset Parkinson's disease 7. This protein belongs to the peptidase C56 family of proteins. It acts as a positive regulator of androgen receptor-dependent transcription. It may also function as a redox-sensitive chaperone, as a sensor for oxidative stress, and it apparently protects neurons against oxidative stress and cell death. -

PARK7 Rabbit Pab
Leader in Biomolecular Solutions for Life Science PARK7 Rabbit pAb Catalog No.: A18580 Basic Information Background Catalog No. The product of this gene belongs to the peptidase C56 family of proteins. It acts as a A18580 positive regulator of androgen receptor-dependent transcription. It may also function as a redox-sensitive chaperone, as a sensor for oxidative stress, and it apparently protects Observed MW neurons against oxidative stress and cell death. Defects in this gene are the cause of 22KDa autosomal recessive early-onset Parkinson disease 7. Two transcript variants encoding the same protein have been identified for this gene. Calculated MW 19kDa Category Primary antibody Applications WB, IHC, IF Cross-Reactivity Human, Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 11315 Q99497 IHC 1:50 - 1:200 Immunogen 1:50 - 1:200 IF Recombinant protein of human PARK7. Synonyms DJ-1;DJ1;GATD2;HEL-S-67p;PARK7 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using PARK7 antibody (A18580) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). Exposure time: 10s. Immunohistochemistry of paraffin- Immunofluorescence analysis of C6 cells Immunofluorescence analysis of HeLa cells embedded human esophageal using using PARK7 Rabbit pAb (A18580) at using PARK7 Rabbit pAb (A18580) at PARK7 Rabbit pAb (A18580) at dilution of dilution of 1:100 (40x lens). -

9 VI June 2021
9 VI June 2021 https://doi.org/10.22214/ijraset.2021.36192 International Journal for Research in Applied Science & Engineering Technology (IJRASET) ISSN: 2321-9653; IC Value: 45.98; SJ Impact Factor: 7.429 Volume 9 Issue VI June 2021- Available at www.ijraset.com Effect of Harmine against Parkinson’s Disease Protein (PARK7) through Molecular Docking Studies Love Kumar1, Komal Kaushik1, Khushboo Rana1, Noopur Khare2, 3, Abhimanyu Kumar Jha1, 2* 1Department of Biotechnology, Faculty of Life Sciences, Institute of Applied Medicines and Research, Ghaziabad, Uttar Pradesh, India. 2Institute of Technology and Management, Meerut, Uttar Pradesh, Affiliated to Dr. A.P.J. Abdul Kalam Technical University, Lucknow, Uttar Pradesh, India. 3Shri Ramswaroop Memorial University, Barabanki, Uttar Pradesh, India. Abstract: Parkinson’s disease (PD) is a common known neurodegenerative disorder with unknown etiology. It was estimated about 0.3% prevalence in the U.S population and enhance to 4 to 5% in older than 85 years. All studies were depending on the molecular docking where all ligands and protein PARK7 (PDB ID: 2RK3) were interacted by docked process. Some natural compounds was selected such as Harmine, Alloxan, Alpha spinasterol, Myrcene, and Vasicinone and PARK7 (PDB ID: 2RK3) protein. According to the PyRx and SWISS ADME result, Harmine was the only ligand which was showing minimum binding affinity. AutoDock Vina software was used for docking process between ligand (Harmine) and receptor protein PARK7 (PDB ID: 2RK3). The result was visualized under PyMol. Harmine was inhibiting the activity of PARK7 (PDB ID: 2RK3) and it may be used for the treatment of PD in future prospect after its in vitro and in vivo studies. -

PARK7 Monoclonal Antibody, Clone 1B11
PARK7 monoclonal antibody, clone Gene Alias: DJ-1, DJ1, FLJ27376, FLJ34360, 1B11 FLJ92274 Gene Summary: The product of this gene belongs to Catalog Number: MAB1076 the peptidase C56 family of proteins. It acts as a positive Regulatory Status: For research use only (RUO) regulator of androgen receptor-dependent transcription. It may also function as a redox-sensitive chaperone, as Product Description: Mouse monoclonal antibody a sensor for oxidative stress, and it apparently protects raised against full length recombinant PARK7. neurons against oxidative stress and cell death. Defects in this gene are the cause of autosomal recessive Clone Name: 1B11 early-onset Parkinson disease 7. Two transcript variants encoding the same protein have been identified for this Immunogen: Recombinant protein corresponding to gene. [provided by RefSeq] amino acids 1-189 of human PARK7. References: Host: Mouse 1. Improved detection of prostate cancer using a magneto-nanosensor assay for serum circulating Reactivity: Human autoantibodies. Xu L, Lee JR, Hao S, Ling XB, Brooks JD, Wang SX, Gambhir SS. PLoS One. 2019 Aug Applications: ELISA, WB-Ce 12;14(8):e0221051. doi: 10.1371/journal.pone.0221051. (See our web site product page for detailed applications eCollection 2019. information) 2. DJ-1 up-regulates glutathione synthesis during Protocols: See our web site at oxidative stress and inhibits A53T alpha-synuclein http://www.abnova.com/support/protocols.asp or product toxicity. Zhou W, Freed CR. J Biol Chem. 2005 Dec page for detailed protocols 30;280(52):43150-8. Epub 2005 Oct 14. 3. Interaction of DJ-1 with Daxx inhibits apoptosis Form: Liquid signal-regulating kinase 1 activity and cell death. -

The Genetics of Parkinson's Disease and Implications for Clinical Practice
G C A T T A C G G C A T genes Review The Genetics of Parkinson’s Disease and Implications for Clinical Practice Jacob Oliver Day 1 and Stephen Mullin 1,2,* 1 Faculty of Health, University of Plymouth, Plymouth PL4 8AA, UK; [email protected] 2 Department of Clinical and Movement Neurosciences, University College London Institute of Neurology, London WC1N 3BG, UK * Correspondence: [email protected] Abstract: The genetic landscape of Parkinson’s disease (PD) is characterised by rare high penetrance pathogenic variants causing familial disease, genetic risk factor variants driving PD risk in a signif- icant minority in PD cases and high frequency, low penetrance variants, which contribute a small increase of the risk of developing sporadic PD. This knowledge has the potential to have a major impact in the clinical care of people with PD. We summarise these genetic influences and discuss the implications for therapeutics and clinical trial design. Keywords: Parkinson’s disease; genetics; precision medicine; clinical trials; monogenic; polygenic 1. Introduction Parkinson’s disease (PD) is a neurodegenerative condition affecting over 6 million people worldwide that is expected to double in prevalence by 2040 [1]. It is characterised by a core set of movement (motor) abnormalities - slowness of movement, muscle rigidity Citation: Day, J.O.; Mullin, S. The and tremor – as well as a number of non-motor features such as constipation, anxiety and Genetics of Parkinson’s Disease and dementia [2]. There is often a prodromal phase of non-motor symptoms which precede Implications for Clinical Practice. motor symptoms by many years [3]. -

Program in Human Neutrophils Fails To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology Anaplasma phagocytophilum , 20 of which you can access for free at: 2005; 174:6364-6372; ; from submission to initial decision 4 weeks from acceptance to publication J Immunol doi: 10.4049/jimmunol.174.10.6364 http://www.jimmunol.org/content/174/10/6364 Insights into Pathogen Immune Evasion Mechanisms: Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils Dori L. Borjesson, Scott D. Kobayashi, Adeline R. Whitney, Jovanka M. Voyich, Cynthia M. Argue and Frank R. DeLeo cites 28 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2005/05/03/174.10.6364.DC1 This article http://www.jimmunol.org/content/174/10/6364.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* • Why • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Insights into Pathogen Immune Evasion Mechanisms: Anaplasma phagocytophilum Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils1 Dori L. -

DJ-1 Mutations Are a Rare Cause of Recessively Inherited Early Onset
1of6 ELECTRONIC LETTER J Med Genet: first published as 10.1136/jmg.2003.011106 on 1 March 2004. Downloaded from DJ-1 mutations are a rare cause of recessively inherited early onset parkinsonism mediated by loss of protein function P J Lockhart, S Lincoln, M Hulihan, J Kachergus, K Wilkes, G Bisceglio, D C Mash, M J Farrer ............................................................................................................................... J Med Genet 2004;41:e22 (http://www.jmedgenet.com/cgi/content/full/41/3/e22). doi: 10.1136/jmg.2003.011106 arkinson’s disease (PD; OMIM #168600) is a common Key points neurodegenerative disorder characterised by bradykine- Psia, resting tremor, muscle rigidity, and postural instability. The pathological features include loss of dopamin- N Mutations in DJ-1/PARK7 were recently identified as a ergic neurones, in particular within the substantia nigra pars cause of early onset parkinsonism (EO-PD). To identify compacta, and eosinophilic, cytoplasmic inclusions termed known and novel DJ-1 mutations and assess their Lewy bodies.1 Although rare, familial forms of parkinsonism frequency, we performed a comprehensive analysis of provide a powerful tool to determine the molecular pathways DJ-1 in 49 EO-PD patients previously excluded for perturbed in idiopathic PD.23 mutations in the parkin gene. Three loci have been associated with autosomal recessive N There were no alterations to DJ-1 in 48 of the 49 cases early onset parkinsonism (EO-PD): parkin (PARK2),4 the as studied. Mutations in DJ-1 appear to be a rare cause of yet unidentified PARK6,5 and DJ-1 (PARK7).6 Loss of Parkin recessively inherited EO-PD, accounting for ,1% of function is the predominant genetic cause of EO-PD in cases with onset prior to 50 years. -
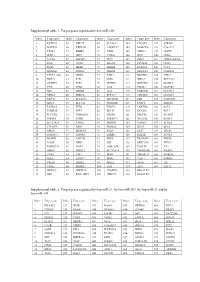
Target Genes Regulated by Hsa-Mir-21, by Hsa-Mir-203, by Hsa-Mir-21 and by Hsa-Mir-143
Supplemental table 1: Target genes regulated by hsa-miR-205 Index Target gene Index Target gene Index Target gene Index Target gene Index Target gene 1 KCTD20 35 UBE2Z 69 SLC38A1 103 LPCAT1 137 STK38L 2 MAPK14 36 YWHAH 70 ANGPTL7 104 MARCKS 138 C1orf123 3 TXNL1 37 RBBP4 71 CTGF 105 MED13 139 GUCD1 4 SPDL1 38 LRP1 72 CYR61 106 IPO7 140 CDK6 5 TCF20 39 IMPAD1 73 TP73 107 PHC2 141 CDKN2AIPNL 6 RAN 40 GNAS 74 EGLN2 108 PICALM 142 CLIP1 7 RGS6 41 MED1 75 ERBB2 109 PLAGL2 143 CUL5 8 HOXA11 42 INPPL1 76 PRRG4 110 NDUFA4 144 C6orf201 9 PAPPA-AS1 43 DDX5 77 F2RL2 111 NDUFB2 145 VTI1A 10 PRR15 44 E2F1 78 GOT1 112 NIPA2 146 SLC5A12 11 ACTRT3 45 E2F5 79 NUFIP2 113 NOTCH2 147 MAML2 12 YES1 46 ZEB2 80 IL24 114 PANK1 148 MAP3K9 13 SRC 47 ERBB3 81 IL32 115 PARD6B 149 NUDT21 14 NPRL3 48 PRKCE 82 RNF217 116 TMEM66 150 DNAJA1 15 NFAT5 49 SLC41A1 83 ZNF585B 117 EZR 151 CCDC108 16 XPOT 50 SLC7A2 84 SIGMAR1 118 ENPP4 152 SHISA6 17 KCTD16 51 ZEB1 85 VEGFA 119 LRRTM4 153 ACP1 18 TMSB4X 52 PHF8 86 BCL9L 120 KCNJ10 154 BCL2 19 PLCXD2 53 TMEM201 87 CREB1 121 PHLPP2 155 NCAPG 20 TNFSF8 54 PTPRJ 88 SERINC3 122 YEATS2 156 KLHL5 21 SLC25A25 55 ETNK1 89 HMGB3 123 VAMP1 157 ACSL4 22 C11orf74 56 XPR1 90 SRD5A1 124 RTN3 158 BCL6 23 GM2A 57 MRPL44 91 PTEN 125 RFX7 159 ITGA5 24 SMNDC1 58 TM9SF2 92 ESRRG 126 RAP2B 160 ACSL1 25 BAMBI 59 PAIP2B 93 PRLR 127 TRAF3IP1 161 EID2B 26 LCOR 60 NEK9 94 ICK 128 SERTAD2 162 TEX35 27 TMEM239 61 NOX5 95 LOH12CR1 129 TOLLIP 163 YY1 28 AMOT 62 DMXL2 96 SLC39A14 130 TMEM55B 164 SMAD1 29 CDK1 63 ETF1 97 BDP1 131 TMEM123 165 SMAD4 30 SQLE 64 -

Dysregulation in the Brain Protein Profile of Zebrafish Lacking the Parkinson’S Disease-Related Protein DJ-1
Molecular Neurobiology (2019) 56:8306–8322 https://doi.org/10.1007/s12035-019-01667-w Dysregulation in the Brain Protein Profile of Zebrafish Lacking the Parkinson’s Disease-Related Protein DJ-1 Amanda J. Edson1 & Helena A. Hushagen1 & Ann Kristin Frøyset1 & Inga Elda1 & Essa A. Khan1 & Antonio Di Stefano2 & Kari E. Fladmark1 Received: 4 February 2019 /Accepted: 31 May 2019 /Published online: 19 June 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract DJ-1 is a protein with a wide range of functions importantly related to redox regulation in the cell. In humans, dysfunction of the PARK7 gene is associated with neurodegeneration and Parkinson’s disease. Our objective was to establish a novel DJ-1 knockout zebrafish line and to identify early brain proteome changes, which could be linked to later pathology. The CRISPR-Cas9 method was used to target exon 1 of the park7-/- gene to produce a transgenic DJ-1-deficient zebrafish model of Parkinson’sdisease. Label-free mass spectrometry was employed to identify altered protein expression in the DJ-1 null brain of early adult animals. The park7−/− line appears to develop normally at young adult and larval stages. With aging however, DJ-1 null fish exhibit lower tyrosine hydroxylase levels, respiratory failure in skeletal muscle, and lower body mass which is especially prevalent among male fish. By proteomic analysis of early adult brains, we determined that less than 5% of the 4091 identified proteins were influenced by the lack of DJ-1. The dysregulated proteins were mainly proteins known to be involved in mitochondrial metabolism, mitophagy, stress response, redox regulation, and inflammation.