Enzymatic Synthesis of Α-Glucosides Using Various Enzymes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Structure of an Acidic Polysaccharide Elaborated by Acetobacter Sp
Agric. Biol. Chem., 50 (5), 1271 ~1278, 1986 1271 Structure of an Acidic Polysaccharide Elaborated by Acetobacter sp. NBI 10051" Kenji Tayama, Hiroyuki Minakami, Seiichi Fujiyama, Hiroshi Masai and Akira Misaki* NakanoBiochemical Research Institute, NakanoVinegar Co., Ltd., Handa, Aichi 475, Japan * Faculty of Science of Living, Osaka City University, Sugimoto-cho, Sumiyoshi, Osaka 558, Japan Received November 19, 1985 An extracellular acidic polysaccharide elaborated by Acetobacter sp. NBI1005 was composed of D-glucose, D-galactose, D-mannose, and D-glucuronic acid (approximate molar ratio, 6 : 2 : 1 : 1). Methylation and fragmentation analysis by partial acid hydrolysis indicated that the polysaccharide has a branched structure containing a backbone chain of /?-(l ->4)-linked D-glucose residues, two out of every four glucose residues being substituted at the 0-3 positions to form two kinds of branches, one consisting of D-mannose and D-glucuronic acid residues and the other of (l ->6)-linked D-galactose and D-glucose residues. Some microorganisms belonging to Ace- cosyl-(l ->4)-D-glucuronosyl-(l ->2)-D-man- tobacter species have been knownto produce nose.9) This paper reports the structural fea- extracellular polysaccharides, such as cellu- ture of polysaccharide AM-1, as revealed by lose,1* dextran,2) levan,3) and an acidic poly- methylation, fragmentation analysis, and en- saccharide,4* and also soluble, /?-(l-»2)- zymatic degradation. branched, £-(1 ^4)-D-glucan5) and )8-(l -»2)- D-glucan.6) In the course of study on acetic MATERIALS AND METHODS acid bacteria (genera Acetobacter and Glu- conobacter) having a high productivity of Materials. Polysaccharide AM-1of Acetobacter sp. -

Improving the Utilization of Isomaltose and Panose by Lager Yeast Saccharomyces Pastorianus
fermentation Article Improving the Utilization of Isomaltose and Panose by Lager Yeast Saccharomyces pastorianus Javier Porcayo Loza 1,2,† , Anna Chailyan 3, Jochen Forster 3 , Michael Katz 3, Uffe Hasbro Mortensen 2,* and Rosa Garcia Sanchez 3,* 1 Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800 Kongens Lyngby, Denmark; [email protected] 2 Department of Bioengineering, Technical University of Denmark, 2800 Kongens Lyngby, Denmark 3 Carlsberg A/S, Carlsberg Research Laboratory, 1799 Copenhagen V, Denmark; [email protected] (A.C.); [email protected] (J.F.); [email protected] (M.K.) * Correspondence: [email protected] (U.H.M.); [email protected] (R.G.S.) † Current address: Graphenea S.A., 20009 San Sebastian, Spain. Abstract: Approximately 25% of all carbohydrates in industrial worts are poorly, if at all, fermented by brewing yeast. This includes dextrins, β-glucans, arabinose, xylose, disaccharides such as isomaltose, nigerose, kojibiose, and trisaccharides such as panose and isopanose. As the efficient utilization of carbohydrates during the wort’s fermentation impacts the alcohol yield and the organoleptic traits of the product, developing brewing strains with enhanced abilities to ferment subsets of these sugars is highly desirable. In this study, we developed Saccharomyces pastorianus laboratory yeast strains with a superior capacity to grow on isomaltose and panose. First, we designed a plasmid toolbox for Citation: Porcayo Loza, J.; Chailyan, the stable integration of genes into lager strains. Next, we used the toolbox to elevate the levels of A.; Forster, J.; Katz, M.; Mortensen, the α-glucoside transporter Agt1 and the major isomaltase Ima1. -

Rational Design of an Improved Transglucosylase for Production of the Rare Sugar Nigerose† Cite This: Chem
ChemComm COMMUNICATION Rational design of an improved transglucosylase for production of the rare sugar nigerose† Cite this: Chem. Commun., 2019, 55,4531 Jorick Franceus,a Shari Dhaene,a Hannes Decadt,a Judith Vandepitte,a Received 25th February 2019, Jurgen Caroen,b Johan Van der Eycken,b Koen Beerensa and Tom Desmet *a Accepted 26th March 2019 DOI: 10.1039/c9cc01587f rsc.li/chemcomm The sucrose phosphorylase from Bifidobacterium adolescentis (BaSP) can be used as a transglucosylase for the production of rare sugars. We designed variants of BaSP for the efficient synthesis of nigerose from sucrose and glucose, thereby adding to the inventory of rare sugars that can conveniently be produced from bulk sugars. Rare sugars hold tremendous potential for practical applications in various industries.1 Regardless, few of them have been exploited Fig. 1 Transglucosylation of glucose by mutant Q345F of the B. adoles- centis sucrose phosphorylase, resulting in the synthesis of maltose and commercially due to their scarcity in nature which prevents nigerose. them from being isolated in large quantities. These compounds have consequently become attractive targets for biocatalytic production processes starting from affordable and widely avail- kojibiose, but instead produces an equimolar mixture of maltose able carbohydrates.2 and nigerose (Fig. 1).9–12 The sucrose phosphorylase from Bifidobacterium adolescentis Nigerose is the rare a-(1,3)-bonded disaccharide of glucose that (BaSP; carbohydrate-active enzyme database family GH13) is a occurs in nature as a constituent of polysaccharides such as nigeran. particularly interesting candidate enzyme for the production of It is also found in Japanese rice wine or sake, hence its alternative such rare sugars. -

Structural Features
1 Structural features As defined by the International Union of Pure and Applied Chemistry gly- cans are structures of multiple monosaccharides linked through glycosidic bonds. The terms sugar and saccharide are synonyms, depending on your preference for Arabic (“sukkar”) or Greek (“sakkēaron”). Saccharide is the root for monosaccha- rides (a single carbohydrate unit), oligosaccharides (3 to 20 units) and polysac- charides (large polymers of more than 20 units). Carbohydrates follow the basic formula (CH2O)N>2. Glycolaldehyde (CH2O)2 would be the simplest member of the family if molecules of two C-atoms were not excluded from the biochemical repertoire. Glycolaldehyde has been found in space in cosmic dust surrounding star-forming regions of the Milky Way galaxy. Glycolaldehyde is a precursor of several organic molecules. For example, reaction of glycolaldehyde with propenal, another interstellar molecule, yields ribose, a carbohydrate that is also the backbone of nucleic acids. Figure 1 – The Rho Ophiuchi star-forming region is shown in infrared light as captured by NASA’s Wide-field Infrared Explorer. Glycolaldehyde was identified in the gas surrounding the star-forming region IRAS 16293-2422, which is is the red object in the centre of the marked square. This star-forming region is 26’000 light-years away from Earth. Glycolaldehyde can react with propenal to form ribose. Image source: www.eso.org/public/images/eso1234a/ Beginning the count at three carbon atoms, glyceraldehyde and dihydroxy- acetone share the common chemical formula (CH2O)3 and represent the smallest carbohydrates. As their names imply, glyceraldehyde has an aldehyde group (at C1) and dihydoxyacetone a carbonyl group (at C2). -

Kalinka Christopher Thesis.Pdf (3.535Mb)
UNIVERSITY OF WISCONSIN-LA CROSSE Graduate Studies DOES THE YICJI OPERON CONTRIBUTE TO UROPATHOGENIC ESCHERICHIA COLI FITNESS WHEN CULTURED IN HUMAN URINE A Manuscript Style Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Microbiology Christopher Kalinka College of Science and Health Microbiology May, 2020 ABSTRACT Kalinka, C.P. Does the yicJI operon contribute to uropathogenic Escherichia coli fitness when cultured in human urine. MS in Microbiology, May 2020, 81pp. (W.Schwan). Escherichia coli is a common bacterium found in the intestinal tracts of many mammals. A medically relevant type of E. coli is known as uropathogenic E. coli (UPEC). One of the most critical research areas involves genes that are responsible for bacterial fitness, like the yicJ gene, which encodes the sodium:galactoside symporter; yicI, which encodes the α-glycosidase YicI; and frzR, which encodes FrzR that is the activator of the frz operon and a putative activator of the yicJI operon. To examine what impacts yicJ, yicI, the yicJI operon, and frzR had on fitness, each gene was cloned into the pMMB91 plasmid and growth curves in urine and buffered Luria broth were done for each recombinant strain in 96-well plates from 0 h-72 h. The growth curve studies demonstrated that overexpression of yicJI and frzR significantly enhanced growth in shaken human urine, whereas overexpression of yicJI attenuated growth in Luria-Bertani broth. Additionally, a portion of the promoter region of frzR was also cloned into the pCRISPathBrick plasmid to act as a gRNA scaffold for dCas9 to knock down the gene expression of the frzR gene. -

(1→3)-Glucan from Laetiporus Sulphureus: a Pilot Study
molecules Communication Prebiotic Potential of Oligosaccharides Obtained by Acid Hydrolysis of α-(1!3)-Glucan from Laetiporus sulphureus: A Pilot Study Adrian Wiater 1,* , Adam Wa´sko 2,*, Paulina Adamczyk 1, Klaudia Gustaw 2, Małgorzata Pleszczy ´nska 1, Kamila Wlizło 1, Marcin Skowronek 3 , Michał Tomczyk 4 and Janusz Szczodrak 1 1 Department of Industrial and Environmental Microbiology, Institute of Biological Science, Maria Curie-Skłodowska University, Akademicka 19, 20-033 Lublin, Poland; [email protected] (P.A.); [email protected] (M.P.); [email protected] (K.W.); [email protected] (J.S.) 2 Department of Biotechnology, Human Nutrition and Food Commodity Science, University of Life Sciences in Lublin, Skromna 8, 20-704 Lublin, Poland; [email protected] 3 Laboratory of Biocontrol, Application and Production of EPN, Centre for Interdisciplinary Research, Faculty of Biotechnology and Environmental Sciences, John Paul II Catholic University of Lublin, ul. Konstantynów 1J, 20-708 Lublin, Poland; [email protected] 4 Department of Pharmacognosy, Faculty of Pharmacy, Medical University of Bialystok, ul. Mickiewicza 2a, 15-230 Białystok, Poland; [email protected] * Correspondence: [email protected] (A.W.); [email protected] (A.W.) Academic Editors: Rossella Grande and Mads Hartvig Clausen Received: 17 September 2020; Accepted: 23 November 2020; Published: 26 November 2020 Abstract: Increasing knowledge of the role of the intestinal microbiome in human health and well-being has resulted in increased interest in prebiotics, mainly oligosaccharides of various origins. To date, there are no reports in the literature on the prebiotic properties of oligosaccharides produced by the hydrolysis of pure fungal α-(1 3)-glucan. -

(12) Patent Application Publication (10) Pub. No.: US 2006/0188465 A1 Perrier Et Al
US 2006O188465A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0188465 A1 Perrier et al. (43) Pub. Date: Aug. 24, 2006 (54) CROSS-LINKED POLYMER OF (30) Foreign Application Priority Data CARBOHYDRATE, NOTABLY BASED ON POLYSACCHARIDES, AND/OR ON Feb. 18, 2005 (FR)............................................ O5 O1674 OLGOSACCHARDES AND/OR ON POLYOLS Publication Classification (75) Inventors: Eric Perrier, Les Cotes D'Arey (FR): (51) Int. Cl. Nabil Abdul-Malak, Caluire (FR): A6IR 8/73 (2006.01) Julie Saget, Lyon (FR) A6II 3/765 (2006.01) C08G 63/9. (2006.01) Correspondence Address: (52) U.S. Cl. ................... 424/70.13; 424/78.37; 525/54.2 MERCHANT & GOULD PC P.O. BOX 2903 (57) ABSTRACT MINNEAPOLIS, MN 55402-0903 (US) (73) Assignee: Engelhard Lyon, Lyon (FR) The invention discloses a cross-linked polymer of at least one carbohydrate component comprising at least one pri (21) Appl. No.: 11/174,414 mary alcohol function in particular for the manufacture of a composition which is intended to be applied onto the skin to (22) Filed: Jul. 1, 2005 obtain a tensing and/or toning effect. US 2006/0188465 A1 Aug. 24, 2006 CROSS-LINKED POLYMER OF CARBOHYDRATE, which be capable of obtaining a tensing and/or toning effect NOTABLY BASED ON POLYSACCHARIDES, on the cutaneous Surface in a way which is perceptible by the AND/OR ON OLGOSACCHARDES AND/OR ON users, without inducing any identifiable allergic reaction. POLYOLS 0010. An aim of the present invention is to solve the 0001. The present invention relates to the preparation of technical problem consisting of providing a toning Solution an active principle having a tensing effect, based on various which is preferably based on a starting material of excellent protein or peptide Substances, so as to avoid problems of biocompatibility, of total biodegradability, of complete allergies encountered with these substances. -
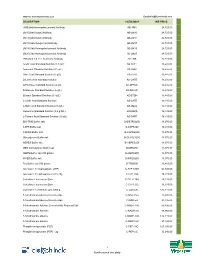
Description Catalog # Inr Price Jim5
Website: www.BiotechIndia.com Email:[email protected] DESCRIPTION CATALOG # INR PRICE JIM5 [Anti-Homogalacturonan] Antibody AB-JIM5 24,725.00 LM10 [Anti-Xylan] Antibody AB-LM10 24,725.00 LM11 [Anti-Xylan] Antibody AB-LM11 24,725.00 LM15 [Anti-Xyloglucan] Antibody AB-LM15 24,725.00 LM19 [Anti-Homogalacturonan] Antibody AB-LM19 24,725.00 LM20 [Anti-Homogalacturonan] Antibody AB-LM20 24,725.00 LM6 [Anti-1,5-α-L-Arabinan] Antibody AB-LM6 24,725.00 Acetic Acid Standard Solution (1.8 g/L) AS-ACET 16,485.00 Ammonia Standard Solution (0.1 g/L) AS-AMIA 16,485.00 Citric Acid Standard Solution (3 g/L) AS-CITR 16,485.00 D-Lactic Acid Standard Solution AS-DATE 16,485.00 D-Fructose Standard Solution (6 g/L AS-DFRUC 16,485.00 D-Glucose Standard Solution (6 g/L) AS-DGLUC 16,485.00 Ethanol Standard Solution (0.3 g/L) AS-ETOH 16,485.00 L-Lactic Acid Standard Solution AS-LATE 16,485.00 L-Malic Acid Standard Solution (6 g/L) AS-LMAL 16,485.00 Isoleucine Standard Solution (0.2 g N/L) AS-NOPA 16,485.00 L-Tartaric Acid Standard Solution (10 g/L) AS-TART 16,485.00 BIS-TRIS Buffer salt B-BISTRIS250 18,070.00 CAPS Buffer salt B-CAPS200 16,485.00 CAPSO Buffer salt B-CAPSO250 18,070.00 Glycylglycine Buffer salt B-GLYGLY250 18,070.00 HEPES Buffer salt B-HEPES250 18,070.00 MES monohydrate Buffer salt B-MES250 18,070.00 MOPS Buffer salt 250 grams B-MOPS250 18,070.00 PIPES Buffer salt B-PIPES250 18,070.00 Tris Buffer salt 500 grams B-TRIS500 16,485.00 Adenosine 5'-triophosphate (ATP) C-ATP-100G 64,345.00 Adenosine 5'-triphosphate (ATP) 20g C-ATP-20G 18,225.00 Calcofluor -

Metabolomics and Transcriptomics to Decipher Molecular Mechanisms Underlying Ectomycorrhizal Root Colonization of an Oak Tree M
www.nature.com/scientificreports OPEN Metabolomics and transcriptomics to decipher molecular mechanisms underlying ectomycorrhizal root colonization of an oak tree M. Sebastiana1*, A. Gargallo‑Garriga2, J. Sardans3,4, M. Pérez‑Trujillo5, F. Monteiro6,7, A. Figueiredo1, M. Maia1,8, R. Nascimento1, M. Sousa Silva8, A. N. Ferreira8, C. Cordeiro8, A. P. Marques8, L. Sousa9, R. Malhó1 & J. Peñuelas3,4,10 Mycorrhizas are known to have a positive impact on plant growth and ability to resist major biotic and abiotic stresses. However, the metabolic alterations underlying mycorrhizal symbiosis are still understudied. By using metabolomics and transcriptomics approaches, cork oak roots colonized by the ectomycorrhizal fungus Pisolithus tinctorius were compared with non‑colonized roots. Results show that compounds putatively corresponding to carbohydrates, organic acids, tannins, long‑ chain fatty acids and monoacylglycerols, were depleted in ectomycorrhizal cork oak colonized roots. Conversely, non‑proteogenic amino acids, such as gamma‑aminobutyric acid (GABA), and several putative defense‑related compounds, including oxylipin‑family compounds, terpenoids and B6 vitamers were induced in mycorrhizal roots. Transcriptomic analysis suggests the involvement of GABA in ectomycorrhizal symbiosis through increased synthesis and inhibition of degradation in mycorrhizal roots. Results from this global metabolomics analysis suggest decreases in root metabolites which are common components of exudates, and in compounds related to root external protective layers which could facilitate plant‑fungal contact and enhance symbiosis. Root metabolic pathways involved in defense against stress were induced in ectomycorrhizal roots that could be involved in a plant mechanism to avoid uncontrolled growth of the fungal symbiont in the root apoplast. Several of the identifed symbiosis‑specifc metabolites, such as GABA, may help to understand how ectomycorrhizal fungi such as P. -

Nutrition Receipe for Building Muscle
NUTRITION RECEIPE FOR BUILDING MUSCLE INGREDIENTS 1. Start with breakfast Breakfast fuels the muscles and maintains them after sleeping for hours without nutrients. 2. After breakfast, time your meals and snacks every three hours (this means 5-6 meals/day) This provides nutrients throughout the day preventing your body from breaking down muscle tissue for energy sources. 3. Think total calories - Protein alone will not put on muscle Protein fills of your stomach quickly causing you to be full but it's lower in calories compared to carbohydrates. 4. Your post workout shake is necessary for building and repairing muscle 5. Hydrate with water to break down food and promote digestion Water also helps circulate nutrients throughout your body. If you are trying to gain weight, add in liquid calories to each meal and snack: drink 100% fruit juice, chocolate milk or shakes. 6. Snack before bed Your body's own natural human growth hormone peaks during this time. To maximize muscle building, take in a big snack full of carbohydrates, proteins and healthy fat like a sandwich, a protein shake with fruit, a breakfast sandwich, rice bowl, a peanut butter and jelly sandwich, cereal or chocolate milk.... 7. Consume food containing muscle building elements: leucine, glutamine and creatine Leucine is the only dietary amino acid that has the capacity to stimulate muscle growth. Foods containing leucine include: beef, beans, nuts, fish, milk. Glutamine is the key to building muscle and keeping muscle during intense training. Glutamine containing dilute foods include: beef, chicken, fish, eggs, milk, dairy products, wheat, cabbage, beets, beans, spinach. -

CYCLOTETRAGLUCOSE and CYCLOTETRAGLUCOSE SYRUP Chemical and Technical Assessment
CYCLOTETRAGLUCOSE AND CYCLOTETRAGLUCOSE SYRUP Chemical and Technical Assessment Prepared by Yoko Kawamura, Ph.D. 1. Summary Cyclotetraglucose is a non-reducing cyclic tetrasaccharide comprised of four D-glucopyranosyl residues linked by alternating α(1→3) and α(1→6) glucosidic bonds. Cyclotetraglucose syrup is a mixture consisting of mainly cyclotetraglucose with branched cyclotetraglucose, mono- and disaccharides, and linear branched or not branched oligosaccharides. They are produced from hydrolyzed food-grade starch by the actions of a mixture of 6-α-glucosyltransferase and α-isomaltosyltransferase derived from Sporosarcina globispora, and cyclodextrin glucosyltransferase derived from Bacillus stearothermophilus from liquefied food-grade starch as the starting material by the joint actions of 6-α-glucosyltransferase and α- isomaltosyltransferase, both isolated from Sporosarcina globispora. Depending on the following steps, crystalline cyclotetraglucose and cyclotetraglucose syrup are produced separately. They are used as a carrier. Cyclotetraglucose had not been evaluated by the Committee and it was placed on the agenda at the request of 37th session of CCFAC (2005) under the name cyclotetraose. The Committee considered that the name cyclotetraose was misleading as it suggests that the substance is a four-carbon sugar whereas it is actually a cyclic tetramer of glucose. The Committee therefore assigned it the name cyclotetraglucose. In reaching its decision the Committee took into account the principles on nomenclature elaborated at its 33rd meeting. 2. Description Cyclotetraglucose is a non-reducing cyclic tetrasaccharide comprised of four D-glucopyranosyl residues linked by alternating α(1→3) and α(1→6) glucosidic bonds. The chemical name is cyclo[→6)-α-D- glucopyranosyl-(1→3)-α-D-glucopyranosyl-(1→6)-α-D-glucopyranosyl-(1→3)- α- D-glucopyranosyl-(1→]. -

M. C. Cam,IUS Northern Regional Research Center, Agricllltural Research Service, U.S
Reprinted From: ADVANCES IN APPLIED MICROBIOLOGY Vol. © 1978, Academic Press, Inc. .N Yorlf. San FrancisCo London Production of Microbial Polysaccl'1anaes M. E. SLODKI A:\D M. C. CAm,IUS Northern Regional Research Center, AgriCllltural Research Service, U.S. Department ofAgriculture! Peoria, Illinois I. Introduction..................................... 19 II. Xanthan Gum. ......................................... 21 A. Background........................................ 21 B. Strain Variability. .................................. 22 C. Production 2.3 D. Other Xanthomonas Polysaccharides 25 III. Arthro!Jacter Polysaccharides. ............................ 26 IV. Microbial Alginic Acids 27 V. Succinoglucan and Curdlan .............................. 29 A. Succinoglucan...................................... 29 B. Curdlan........................................... 31 C. Other ,B-(1~3)-Linked Glucans .. ..................... 32 VI. Other Bacterial Polysaccharides 33 VII. Phosphomannans and ~Iannans. .......................... 34 VIII. Cryptococcus Heteropolysaccharide ....................... 37 L'\. "Black Yeast" Polysaccharides. ........................... 38 X. Pullulan............................................... 40 XI. Polysaccharides from Hydrocarbons and Low-Molecular-Weight Petrochemical Substrates. .. .. ...... 44 A. n-Alkanes 44 B. Lower Polyhydric Alcohols. .......................... 45 C. Methanol.......................................... 46 XII. Concluding Remarks. ................................... 47 References 49 I. Introduction