Meta-Analysis of Bovine Digital Dermatitis Microbiota Reveals Distinct Microbial Community Structures Associated with Lesions
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Macellibacteroides Fermentans Gen. Nov., Sp. Nov., a Member of the Family Porphyromonadaceae Isolated from an Upflow Anaerobic Filter Treating Abattoir Wastewaters
International Journal of Systematic and Evolutionary Microbiology (2012), 62, 2522–2527 DOI 10.1099/ijs.0.032508-0 Macellibacteroides fermentans gen. nov., sp. nov., a member of the family Porphyromonadaceae isolated from an upflow anaerobic filter treating abattoir wastewaters Linda Jabari,1,2 Hana Gannoun,2 Jean-Luc Cayol,1 Abdeljabbar Hedi,1 Mitsuo Sakamoto,3 Enevold Falsen,4 Moriya Ohkuma,3 Moktar Hamdi,2 Guy Fauque,1 Bernard Ollivier1 and Marie-Laure Fardeau1 Correspondence 1Aix-Marseille Universite´ du Sud Toulon-Var, CNRS/INSU, IRD, MIO, UM 110, Case 925, Marie-Laure Fardeau 163 Avenue de Luminy, 13288 Marseille Cedex 9, France [email protected] 2Laboratoire d’Ecologie et de Technologie Microbienne, Institut National des Sciences Applique´es et de Technologie, Centre Urbain Nord, BP 676, 1080 Tunis Cedex, Tunisia 3Microbe Division/Japan Collection of Microorganisms, RIKEN BioResource Center 2-1 Hirosawa, Wako, Saitama 351-0198, Japan 4CCUG, Culture Collection, Department of Clinical Bacteriology, University of Go¨teborg, 41346 Go¨teborg, Sweden A novel obligately anaerobic, non-spore-forming, rod-shaped mesophilic bacterium, which stained Gram-positive but showed the typical cell wall structure of Gram-negative bacteria, was isolated from an upflow anaerobic filter treating abattoir wastewaters in Tunisia. The strain, designated LIND7HT, grew at 20–45 6C (optimum 35–40 6C) and at pH 5.0–8.5 (optimum pH 6.5–7.5). It did not require NaCl for growth, but was able to grow in the presence of up to 2 % NaCl. Sulfate, thiosulfate, elemental sulfur, sulfite, nitrate and nitrite were not used as terminal electron acceptors. -

Comparative Genomics of the Genus Porphyromonas Identifies Adaptations for Heme Synthesis Within the Prevalent Canine Oral Species Porphyromonas Cangingivalis
GBE Comparative Genomics of the Genus Porphyromonas Identifies Adaptations for Heme Synthesis within the Prevalent Canine Oral Species Porphyromonas cangingivalis Ciaran O’Flynn1,*, Oliver Deusch1, Aaron E. Darling2, Jonathan A. Eisen3,4,5, Corrin Wallis1,IanJ.Davis1,and Stephen J. Harris1 1 The WALTHAM Centre for Pet Nutrition, Waltham-on-the-Wolds, United Kingdom Downloaded from 2The ithree Institute, University of Technology Sydney, Ultimo, New South Wales, Australia 3Department of Evolution and Ecology, University of California, Davis 4Department of Medical Microbiology and Immunology, University of California, Davis 5UC Davis Genome Center, University of California, Davis http://gbe.oxfordjournals.org/ *Corresponding author: E-mail: ciaran.ofl[email protected]. Accepted: November 6, 2015 Abstract Porphyromonads play an important role in human periodontal disease and recently have been shown to be highly prevalent in canine mouths. Porphyromonas cangingivalis is the most prevalent canine oral bacterial species in both plaque from healthy gingiva and at University of Technology, Sydney on January 17, 2016 plaque from dogs with early periodontitis. The ability of P. cangingivalis to flourish in the different environmental conditions char- acterized by these two states suggests a degree of metabolic flexibility. To characterize the genes responsible for this, the genomes of 32 isolates (including 18 newly sequenced and assembled) from 18 Porphyromonad species from dogs, humans, and other mammals were compared. Phylogenetic trees inferred using core genes largely matched previous findings; however, comparative genomic analysis identified several genes and pathways relating to heme synthesis that were present in P. cangingivalis but not in other Porphyromonads. Porphyromonas cangingivalis has a complete protoporphyrin IX synthesis pathway potentially allowing it to syn- thesize its own heme unlike pathogenic Porphyromonads such as Porphyromonas gingivalis that acquire heme predominantly from blood. -

The Mysterious Orphans of Mycoplasmataceae
The mysterious orphans of Mycoplasmataceae Tatiana V. Tatarinova1,2*, Inna Lysnyansky3, Yuri V. Nikolsky4,5,6, and Alexander Bolshoy7* 1 Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, 90027, California, USA 2 Spatial Science Institute, University of Southern California, Los Angeles, 90089, California, USA 3 Mycoplasma Unit, Division of Avian and Aquatic Diseases, Kimron Veterinary Institute, POB 12, Beit Dagan, 50250, Israel 4 School of Systems Biology, George Mason University, 10900 University Blvd, MSN 5B3, Manassas, VA 20110, USA 5 Biomedical Cluster, Skolkovo Foundation, 4 Lugovaya str., Skolkovo Innovation Centre, Mozhajskij region, Moscow, 143026, Russian Federation 6 Vavilov Institute of General Genetics, Moscow, Russian Federation 7 Department of Evolutionary and Environmental Biology and Institute of Evolution, University of Haifa, Israel 1,2 [email protected] 3 [email protected] 4-6 [email protected] 7 [email protected] 1 Abstract Background: The length of a protein sequence is largely determined by its function, i.e. each functional group is associated with an optimal size. However, comparative genomics revealed that proteins’ length may be affected by additional factors. In 2002 it was shown that in bacterium Escherichia coli and the archaeon Archaeoglobus fulgidus, protein sequences with no homologs are, on average, shorter than those with homologs [1]. Most experts now agree that the length distributions are distinctly different between protein sequences with and without homologs in bacterial and archaeal genomes. In this study, we examine this postulate by a comprehensive analysis of all annotated prokaryotic genomes and focusing on certain exceptions. -

Correlation Between the Oral Microbiome and Brain Resting State Connectivity in Smokers
bioRxiv preprint doi: https://doi.org/10.1101/444612; this version posted October 16, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Correlation between the oral microbiome and brain resting state connectivity in smokers Dongdong Lin1, Kent Hutchison2, Salvador Portillo3, Victor Vegara1, Jarod Ellingson2, Jingyu Liu1,3, Amanda Carroll-Portillo3,* ,Vince D. Calhoun1,3,* 1The Mind Research Network, Albuquerque, New Mexico, 87106 2University of Colorado Boulder, Boulder, CO 3University of New Mexico, Department of Electrical and Computer Engineering, Albuquerque, New Mexico, 87106 * authors contributed equally to the work. Abstract Recent studies have shown a critical role for the gastrointestinal microbiome in brain and behavior via a complex gut–microbiome–brain axis, however, the influence of the oral microbiome in neurological processes is much less studied, especially in response to the stimuli in the oral microenvironment such as smoking. Additionally, given the complex structural and functional networks in brain system, our knowledge about the relationship between microbiome and brain functions on specific brain circuits is still very limited. In this pilot work, we leverage next generation microbial sequencing with functional MRI techniques to enable the delineation of microbiome-brain network links as well as their relations to cigarette smoking. Thirty smokers and 30 age- and sex- matched non-smokers were recruited for measuring both microbial community and brain functional networks. Statistical analyses were performed to demonstrate the influence of smoking on: the taxonomy and abundance of the constituents within the oral microbial community, brain functional network connectivity, and associations between microbial shifts and the brain signaling network. -

Description of Gabonibacter Massiliensis Gen. Nov., Sp. Nov., a New Member of the Family Porphyromonadaceae Isolated from the Human Gut Microbiota
Curr Microbiol DOI 10.1007/s00284-016-1137-2 Description of Gabonibacter massiliensis gen. nov., sp. nov., a New Member of the Family Porphyromonadaceae Isolated from the Human Gut Microbiota 1,2 1 3,4 Gae¨l Mourembou • Jaishriram Rathored • Jean Bernard Lekana-Douki • 5 1 1 Ange´lique Ndjoyi-Mbiguino • Saber Khelaifia • Catherine Robert • 1 1,6 1 Nicholas Armstrong • Didier Raoult • Pierre-Edouard Fournier Received: 9 June 2016 / Accepted: 8 September 2016 Ó Springer Science+Business Media New York 2016 Abstract The identification of human-associated bacteria Gabonibacter gen. nov. and the new species G. mas- is very important to control infectious diseases. In recent siliensis gen. nov., sp. nov. years, we diversified culture conditions in a strategy named culturomics, and isolated more than 100 new bacterial Keywords Gabonibacter massiliensis Á Taxonogenomics Á species and/or genera. Using this strategy, strain GM7, a Culturomics Á Gabon Á Gut microbiota strictly anaerobic gram-negative bacterium was recently isolated from a stool specimen of a healthy Gabonese Abbreviations patient. It is a motile coccobacillus without catalase and CSUR Collection de Souches de l’Unite´ des oxidase activities. The genome of Gabonibacter mas- Rickettsies siliensis is 3,397,022 bp long with 2880 ORFs and a G?C DSM Deutsche Sammlung von content of 42.09 %. Of the predicted genes, 2,819 are Mikroorganismen protein-coding genes, and 61 are RNAs. Strain GM7 differs MALDI-TOF Matrix-assisted laser desorption/ from the closest genera within the family Porphyromon- MS ionization time-of-flight mass adaceae both genotypically and in shape and motility. -

Endosymbiont Diversity and Evolution Across Weevil Tree of Life
bioRxiv preprint doi: https://doi.org/10.1101/171181; this version posted August 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Endosymbiont diversity and evolution across weevil tree of life Guanyang Zhang1#, Patrick Browne1,2#, Geng Zhen1#, Andrew Johnston4, Hinsby Cadillo-Quiroz5, and Nico Franz1 1 School of Life Sciences, Arizona State University, Tempe, Arizona, USA 2 Department of Environmental Science, Aarhus University, 4000 Roskilde, Denmark # These authors contributed equally to this work ABSTRACT As early as the time of Paul Buchner, a pioneer of endosymbionts research, it was shown that weevils host diverse bacterial endosymbionts, probably only second to the hemipteran insects. To date, there is no taxonomically broad survey of endosymbionts in weevils, which preclude any systematic understanding of the diversity and evolution of endosymbionts in this large group of insects, which comprise nearly 7% of described diversity of all insects. We gathered the largest known taxonomic sample of weevils representing four families and 17 subfamilies to perform a study of weevil endosymbionts. We found that the diversity of endosymbionts is exceedingly high, with as many as 44 distinct kinds of endosymbionts detected. We recovered an ancient origin of association of Nardonella with weevils, dating back to 124 MYA. We found repeated losses of this endosymbionts, but also cophylogeny with weevils. We also investigated patterns of coexistence and coexclusion. INTRODUCTION Weevils (Insecta: Coleoptera: Curculionoidea) host diverse bacterial endosymbionts. -

Research Article Review Jmb
J. Microbiol. Biotechnol. (2017), 27(0), 1–7 https://doi.org/10.4014/jmb.1707.07027 Research Article Review jmb Methods 20,546 sequences and all the archaeal datasets were normalized to 21,154 sequences by the “sub.sample” Bioinformatics Analysis command. The filtered sequences were classified against The raw read1 and read2 datasets was demultiplexed by the SILVA 16S reference database (Release 119) using a trimming the barcode sequences with no more than 1 naïve Bayesian classifier built in Mothur with an 80% mismatch. Then the sequences with the same ID were confidence score [5]. Sequences passing through all the picked from the remaining read1 and read2 datasets by a filtration were also clustered into OTUs at 6% dissimilarity self-written python script. Bases with average quality score level. Then a “classify.otu” function was utilized to assign lower than 25 over a 25 bases sliding window were the phylogenetic information to each OTU. excluded and sequences which contained any ambiguous base or had a final length shorter than 200 bases were Reference abandoned using Sickle [1]. The paired reads were assembled into contigs and any contigs with an ambiguous 1. Joshi NA, FJ. 2011. Sickle: A sliding-window, adaptive, base, more than 8 homopolymeric bases and fewer than 10 quality-based trimming tool for FastQ files (Version 1.33) bp overlaps were culled. After that, the contigs were [Software]. further trimmed to get rid of the contigs that have more 2. Schloss PD. 2010. The Effects of Alignment Quality, than 1 forward primer mismatch and 2 reverse primer Distance Calculation Method, Sequence Filtering, and Region on the Analysis of 16S rRNA Gene-Based Studies. -

I RESEARCH DISSERTATION for Msc Degree in Life Sciences Submitted
INVESTIGATION OF TICK-BORNE PATHOGENS RESISTANCE MARKERS USING NEXT GENERATION SEQUENCING RESEARCH DISSERTATION for MSc degree in Life Sciences Submitted to the DEPARTMENT OF LIFE AND CONSUMER SCIENCES UNIVERSITY OF SOUTH AFRICA by AUBREY D CHIGWADA Student No: 61366943 SUPERVISOR: DR TM MASEBE CO-SUPERVISOR: DR NO MAPHOLI JULY 2021 i DECLARATION Name: Aubrey Dickson Chigwada Student number: 61366943 Degree: Master of Science in Life Sciences Dissertation title: INVESTIGATION OF TICK-BORNE PATHOGENS RESISTANCE MARKERS USING NEXT GENERATION SEQUENCING I, Aubrey D Chigwada, declare that investigation of tick-borne pathogens resistance markers using next-generation sequencing is my work, and sources I have used have been acknowledged by complete references. I submit this thesis for a Master of Science in life science at the college of agriculture and environmental science at the University of South Africa and have not been submitted to any other university. JULY 2021 -------------------------------- --------------------------- Aubrey Dickson Chigwada Date ii ACKNOWLEDGEMENTS Firstly, I would like to thank God almighty for the strength to carry on and a gift of life. Words cannot express my deepest gratitude to my supervisor Dr. TM Masebe for her professional guidance, encouragement, support, corrections, instructions, patience, and technical skills thought- out this work. I am forever indebted to your mentorship; you are a role model and an inspiration to me. I would like to extend my sincere appreciation to my Co-supervisor Dr. NO Mapholi for her insightful comments and suggestions. To the woman in research funds used for this project, I am grateful. Much gratitude to Dr. Ramganesh Selvarajan for his mentorship and training from the basics to the actual Miseq sequencing, which made it possible to sequence for this work. -
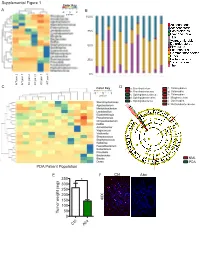
Mental Figure 1 Color Key a -2 0 2 B Z-Score 100%
Supplemental Figure 1 Color Key A -2 0 2 B z-score 100% 75% 50% 25% 0% KC pan 1 WT pan 3 WT KC pan 3 WT pan 2 WT pan 1 WT KC pan 2 C Color Key D a: Brevibacterium f: Chlamydiales b: Brevibacteriaceae g: Chlamydiia -3 0 3 z-score c: Sphingobacteriaceae h: Chlamydiae d: Sphingobacteriales i: Mogibacterium e: Sphingobacteriia j: Oscillospira k: Methylobacteriaceae NML Control Microb.-entrained MΦ PDA PDA Patient Population Control Microb.-entrained MΦ + Myd88i E F Ctrl Abx 350 * 300 250 200 150 40X 100 Tumor weight (mg) 50 0 x Ctrl Ab Supplemental Figure 2 A KC WT B ** * Actinobacteria * ** Bacteroidetes Cyanobacteria Deferribacteres * Firmicutes Proteobacteria % Relative abundance TM7 Others Time(wks) 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 Alpha Diversity Measure C E 60 KC WT 40 20 B. pseudolongum B. animalis 60 5 KC WT 0 B. adolescentis 40% Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 20 Age (weeks) B. pseudolongum B. animalis 5 0 B. adolescentis % Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 F Age (weeks) Week 3 Week 9 Week 13 p=0.678 p=0.02 p=0.385 Time(wks) 3 9 24 20 13 16 D 28 32 36 Week 13 KC WT Firmicutes; Ruminococcus Firmicutes; Dehalobacterium Alpha Diversity Measure Firmicutes; Oscillospira Bacteroidates; Odoribacter Axis.2 [12.7%] Actinobacteria; Bifidobacterium Axis.2 [23.8%] Axis.2 [24.7%] Week 16 Bacteroidetes; Bacteroidales Axis.1 [80.8%] Axis.1 [65.4%] Axis.1 [49.6%] Actinobacteria; Bifidobacterium Week 16 Week 20 Week 24 Week 20 p=0.339 p=0.036 p=0.021 Firmicutes; Dehalobacterium -

Exploring the Cockatiel (Nymphicus Hollandicus) Fecal Microbiome, Bacterial Inhabitants of a Worldwide Pet
Exploring the cockatiel (Nymphicus hollandicus) fecal microbiome, bacterial inhabitants of a worldwide pet Luis David Alcaraz1, Apolinar M. Hernández2 and Mariana Peimbert2 1 Laboratorio Nacional de Ciencias de la Sostenibilidad, Instituto de Ecología, Universidad Nacional Autonóma de México, Mexico City, Mexico 2 Departamento de Ciencias Naturales, Unidad Cuajimalpa, Universidad Autónoma Metropolitana, Mexico City, Mexico ABSTRACT Background. Cockatiels (Nymphicus hollandicus) were originally endemic to Australia; now they are popular pets with a global distribution. It is now possible to conduct detailed molecular studies on cultivable and uncultivable bacteria that are part of the intestinal microbiome of healthy animals. These studies show that bacteria are an essential part of the metabolic capacity of animals. There are few studies on bird microbiomes and, to the best of our knowledge, this is the first report on the cockatiel microbiome. Methods. In this paper, we analyzed the gut microbiome from fecal samples of three healthy adult cockatiels by massive sequencing of the 16S rRNA gene. Additionally, we compared the cockatiel fecal microbiomes with those of other bird species, including poultry and wild birds. Results. The vast majority of the bacteria found in cockatiels were Firmicutes, while Proteobacteria and Bacteroidetes were poorly represented. A total of 19,280 different OTUs were detected, of which 8,072 belonged to the Erysipelotrichaceae family. Discussion. It is relevant to study cockatiel the microbiomes of cockatiels owing to their wide geographic distribution and close human contact. This study serves as a reference for cockatiel bacterial diversity. Despite the large OTU numbers, the diversity is not even Submitted 14 July 2016 Accepted 28 November 2016 and is dominated by Firmicutes of the Erysipelotrichaceae family. -

Prevalence of Ureaplasma Urealyticum, Mycoplasma Hominis and Chlamydia Trachomatis in Patients with Uncomplicated Recurrent Urin
Nephrology and Renal Diseases Research Article ISSN: 2399-908X Prevalence of Ureaplasma urealyticum, Mycoplasma hominis and Chlamydia trachomatis in patients with uncomplicated recurrent urinary tract infections Jadranka Vlasic-Matas1*, Hrvoje Raos2, Marijana Vuckovic2, Stjepan Radic2 and Vesna Capkun3 1Polyclinic Nephrology Department, Split, Croatia 2School of Medicine, University of Split, Split, Croatia 3Department of Nuclear Medicine, Split University Hospital Center, Split, Croatia Abstract Aim: To assess the prevalence of Ureaplasma urealyticum, Mycoplasma hominis and Chlamydia trachomatis in patients with chronic urinary tract infections (UTIs) and its correlation with leukocyturia and symptoms. Methods: The study included 220 patients (130 women and 90 men) presenting with chronic voiding symptoms and sterile leukocyturia. Urine, urethral swabs and cervical swabs (for women patients) were taken to determine the presence of these pathogens. Patients were treated by tetracycline and followed up three and six months after initial therapy. Results: In 186 (85%) out of 220 patients, U. urealyticum was found, while C. trachomatis was present in 34 patients (15%). In majority of female patients (112 out of 130; 86%) U. urealyticum was found. In addition to ureaplasma, in eight patients M. hominis was found. C. trachomatis was identified in 18 female patients (14%). In 74 out of 90 (82%) male patients U. urealyticum was detected while in six of them M. hominis was also found. C. trachomatis was identified in 16 male patients (18%). U. urealyticum was significantly related to leukocyturia, as opposed to C. trachomatis (p<0,001). Women had more frequent symptomatology (p = 0,015) and higer leukocyturia (p<0.001). Conclusion: Leukocyturia is more common find in U.