Mapping of Qtls Detected in a Broccoli Double Diploid Population for Plant Height and Leaf Characteristics
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Genome-Wide Association Study for Circulating Fibroblast Growth Factor
www.nature.com/scientificreports OPEN Genome‑wide association study for circulating fbroblast growth factor 21 and 23 Gwo‑Tsann Chuang1,2,14, Pi‑Hua Liu3,4,14, Tsui‑Wei Chyan5, Chen‑Hao Huang5, Yu‑Yao Huang4,6, Chia‑Hung Lin4,7, Jou‑Wei Lin8, Chih‑Neng Hsu8, Ru‑Yi Tsai8, Meng‑Lun Hsieh9, Hsiao‑Lin Lee9, Wei‑shun Yang2,9, Cassianne Robinson‑Cohen10, Chia‑Ni Hsiung11, Chen‑Yang Shen12,13 & Yi‑Cheng Chang2,9,12* Fibroblast growth factors (FGFs) 21 and 23 are recently identifed hormones regulating metabolism of glucose, lipid, phosphate and vitamin D. Here we conducted a genome‑wide association study (GWAS) for circulating FGF21 and FGF23 concentrations to identify their genetic determinants. We enrolled 5,000 participants from Taiwan Biobank for this GWAS. After excluding participants with diabetes mellitus and quality control, association of single nucleotide polymorphisms (SNPs) with log‑transformed FGF21 and FGF23 serum concentrations adjusted for age, sex and principal components of ancestry were analyzed. A second model additionally adjusted for body mass index (BMI) and a third model additionally adjusted for BMI and estimated glomerular fltration rate (eGFR) were used. A total of 4,201 participants underwent GWAS analysis. rs67327215, located within RGS6 (a gene involved in fatty acid synthesis), and two other SNPs (rs12565114 and rs9520257, located between PHC2-ZSCAN20 and ARGLU1-FAM155A respectively) showed suggestive associations with serum FGF21 level (P = 6.66 × 10–7, 6.00 × 10–7 and 6.11 × 10–7 respectively). The SNPs rs17111495 and rs17843626 were signifcantly associated with FGF23 level, with the former near PCSK9 gene and the latter near HLA-DQA1 gene (P = 1.04 × 10–10 and 1.80 × 10–8 respectively). -

PHC1 Maintains Pluripotency by Organizing Genome-Wide Chromatin Interactions of the Nanog Locus
ARTICLE https://doi.org/10.1038/s41467-021-22871-0 OPEN PHC1 maintains pluripotency by organizing genome-wide chromatin interactions of the Nanog locus Li Chen1,2,3,14, Qiaoqiao Tong1,2,3,14, Xiaowen Chen4,14, Penglei Jiang1,2, Hua Yu1,2, Qianbing Zhao1,2,3, Lingang Sun1,2,3, Chao Liu 1,2,3,5, Bin Gu6, Yuping Zheng 7, Lijiang Fei1,2,3, Xiao Jiang8, Wenjuan Li9,10, Giacomo Volpe 9,10, Mazid MD. Abdul9,10, Guoji Guo 1,2,3, Jin Zhang1,2, Pengxu Qian1,2, Qiming Sun 8, ✉ ✉ ✉ Dante Neculai 7, Miguel A. Esteban9,10,11, Chen Li 12 , Feiqiu Wen4 & Junfeng Ji 1,2,3,13 1234567890():,; Polycomb group (PcG) proteins maintain cell identity by repressing gene expression during development. Surprisingly, emerging studies have recently reported that a number of PcG proteins directly activate gene expression during cell fate determination process. However, the mechanisms by which they direct gene activation in pluripotency remain poorly under- stood. Here, we show that Phc1, a subunit of canonical polycomb repressive complex 1 (cPRC1), can exert its function in pluripotency maintenance via a PRC1-independent activa- tion of Nanog. Ablation of Phc1 reduces the expression of Nanog and overexpression of Nanog partially rescues impaired pluripotency caused by Phc1 depletion. We find that Phc1 interacts with Nanog and activates Nanog transcription by stabilizing the genome-wide chromatin interactions of the Nanog locus. This adds to the already known canonical function of PRC1 in pluripotency maintenance via a PRC1-dependent repression of differentiation genes. Overall, our study reveals a function of Phc1 to activate Nanog transcription through regulating chromatin architecture and proposes a paradigm for PcG proteins to maintain pluripotency. -

Juxtaposed Polycomb Complexes Co-Regulate Vertebral Identity
RESEARCH ARTICLE 4957 Development 133, 4957-4968 (2006) doi:10.1242/dev.02677 Juxtaposed Polycomb complexes co-regulate vertebral identity Se Young Kim1, Suzanne W. Paylor1, Terry Magnuson2 and Armin Schumacher1,* Best known as epigenetic repressors of developmental Hox gene transcription, Polycomb complexes alter chromatin structure by means of post-translational modification of histone tails. Depending on the cellular context, Polycomb complexes of diverse composition and function exhibit cooperative interaction or hierarchical interdependency at target loci. The present study interrogated the genetic, biochemical and molecular interaction of BMI1 and EED, pivotal constituents of heterologous Polycomb complexes, in the regulation of vertebral identity during mouse development. Despite a significant overlap in dosage-sensitive homeotic phenotypes and co-repression of a similar set of Hox genes, genetic analysis implicated eed and Bmi1 in parallel pathways, which converge at the level of Hox gene regulation. Whereas EED and BMI1 formed separate biochemical entities with EzH2 and Ring1B, respectively, in mid-gestation embryos, YY1 engaged in both Polycomb complexes. Strikingly, methylated lysine 27 of histone H3 (H3-K27), a mediator of Polycomb complex recruitment to target genes, stably associated with the EED complex during the maintenance phase of Hox gene repression. Juxtaposed EED and BMI1 complexes, along with YY1 and methylated H3- K27, were detected in upstream regulatory regions of Hoxc8 and Hoxa5. The combined data suggest a model wherein epigenetic and genetic elements cooperatively recruit and retain juxtaposed Polycomb complexes in mammalian Hox gene clusters toward co- regulation of vertebral identity. KEY WORDS: Polycomb, eed, Bmi1, Hox genes, Mouse development, Chromatin, Histones, Epigenetics INTRODUCTION Wang, H. -

A High-Density Map for Navigating the Human Polycomb Complexome
Resource A High-Density Map for Navigating the Human Polycomb Complexome Graphical Abstract Authors Simon Hauri, Federico Comoglio, Makiko Seimiya, ..., Renato Paro, Matthias Gstaiger, Christian Beisel Correspondence [email protected] (M.G.), [email protected] (C.B.) In Brief Polycomb group (PcG) proteins mediate gene silencing and epigenetic memory in higher eukaryotes. By systematically mapping the human PcG complexome, Hauri et al. resolve Polycomb subcomplexes at high resolution and identify two human PRC2 and two PR- DUB complexes. Furthermore, genomic profiling reveals segregation of PRC1 and PR-DUB target genes. Highlights Accession Numbers d 1,400 high-confidence interactions reveal the modular GSE51673 organization of human PcG proteins d Detailed dissection of PRC1 and PRC2 subcomplexes d Two human PR-DUB complexes contain the glycosyltransferase OGT1 d PR-DUB and PRC1 bind largely distinct sets of target genes Hauri et al., 2016, Cell Reports 17, 583–595 October 4, 2016 ª 2016 The Authors. http://dx.doi.org/10.1016/j.celrep.2016.08.096 Cell Reports Resource A High-Density Map for Navigating the Human Polycomb Complexome Simon Hauri,1,2,7,8 Federico Comoglio,3,7,9 Makiko Seimiya,3 Moritz Gerstung,3,10 Timo Glatter,1,11 Klaus Hansen,4 Ruedi Aebersold,1,5 Renato Paro,3,6 Matthias Gstaiger,1,2,* and Christian Beisel3,12,* 1Department of Biology, Institute of Molecular Systems Biology, ETH Zurich,€ 8093 Zurich,€ Switzerland 2Competence Center Personalized Medicine UZH/ETH, 8044 Zurich,€ Switzerland 3Department -

Differential DNA Methylation Patterns Define Status Epilepticus and Epileptic Tolerance
The Journal of Neuroscience, February 1, 2012 • 32(5):1577–1588 • 1577 Neurobiology of Disease Differential DNA Methylation Patterns Define Status Epilepticus and Epileptic Tolerance Suzanne F. C. Miller-Delaney,1 Sudipto Das,2,4 Takanori Sano,1 Eva M. Jimenez-Mateos,1 Kenneth Bryan,2,4 Patrick G. Buckley,2,3,4 Raymond L. Stallings,2,4 and David C. Henshall1 Departments of 1Physiology and Medical Physics and 2Cancer Genetics and 3Molecular and Cellular Therapeutics, Royal College of Surgeons in Ireland, Dublin 2, Ireland, and 4National Children’s Research Centre, Our Lady’s Children’s Hospital, Dublin 12, Ireland Prolonged seizures (status epilepticus) produce pathophysiological changes in the hippocampus that are associated with large-scale, wide-ranging changes in gene expression. Epileptic tolerance is an endogenous program of cell protection that can be activated in the brainbypreviousexposuretoanon-harmfulseizureepisodebeforestatusepilepticus.Amajortranscriptionalfeatureoftoleranceisgene downregulation. Here, through methylation analysis of 34,143 discrete loci representing all annotated CpG islands and promoter regions in the mouse genome, we report the genome-wide DNA methylation changes in the hippocampus after status epilepticus and epileptic tolerance in adult mice. A total of 321 genes showed altered DNA methylation after status epilepticus alone or status epilepticus that followed seizure preconditioning, with Ͼ90% of the promoters of these genes undergoing hypomethylation. These profiles included genes not previously associated with epilepsy, such as the polycomb gene Phc2. Differential methylation events generally occurred throughout the genome without bias for a particular chromosomal region, with the exception of a small region of chromosome 4, which was significantly overrepresented with genes hypomethylated after status epilepticus. -

Sterile Alpha Motif Containing 7 (Samd7) Is a Novel Crx- Regulated Transcriptional Repressor in the Retina
Sterile Alpha Motif Containing 7 (Samd7) Is a Novel Crx- Regulated Transcriptional Repressor in the Retina Julia Hlawatsch1., Marcus Karlstetter1,2., Alexander Aslanidis1,2., Anika Lu¨ ckoff2, Yana Walczak1,2, Michael Plank1, Julia Bo¨ ck1, Thomas Langmann1,2* 1 Institute of Human Genetics, University of Regensburg, Regensburg, Germany, 2 Department of Ophthalmology, University of Cologne, Cologne, Germany Abstract Inherited retinal diseases are mainly caused by mutations in genes that are highly expressed in photoreceptors of the retina. The majority of these genes is under the control of the transcription factor Cone rod homeobox (Crx), that acts as a master transcription factor in photoreceptors. Using a genome-wide chromatin immunoprecipitation dataset that highlights all potential in vivo targets of Crx, we have identified a novel sterile alpha motif (SAM) domain containing protein, Samd7. mRNA Expression of Samd7 was confined to the late postnatal and adult mouse retina as well as the pineal gland. Using immunohistochemistry and Western blot, we could detect Samd7 protein in the outer nuclear layer of adult mouse retina. Ectopic over-expression in HEK293 cells demonstrated that Samd7 resides in the cytoplasm as well as the nucleus. In vitro electroporation of fluorescent reporters into living mouse retinal cultures revealed that transcription of the Samd7 gene depends on evolutionary conserved Crx motifs located in the first intron enhancer. Moreover, Crx knock-down with shRNA strongly reduced Samd7 reporter activity and endogenous Samd7 protein, indicating that Crx is required for retinal expression of Samd7. Finally, using co-transfections in luciferase reporter assays we found that Samd7 interferes with Crx- dependent transcription. -

PTE, a Novel Module to Target Polycomb Repressive Complex 1 to the Cyclin D2
bioRxiv preprint doi: https://doi.org/10.1101/177097; this version posted October 23, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. PTE, a novel module to target Polycomb Repressive Complex 1 to the Cyclin D2 oncogene Sarina R. Cameron1, Soumyadeep Nandi1,2,¤, Tatyana G. Kahn1,¤, Juan I. Barrasa1,¤, Per Stenberg1,2,3, Yuri B. Schwartz1,# 1 Department of Molecular Biology, Umeå University, Umeå, Sweden 2 Computational Life Science Cluster (CLiC), Umeå University, Umeå, Sweden 3 Division of CBRN Security and Defence, FOI–Swedish Defence Research Agency, Umeå, Sweden #correspondence should be addressed to: [email protected] ¤joint second authors Running title: PRC1 targeting element Key words: Polycomb/ Polycomb targeting/ PRC1/ epigenetics/ Cyclin D2 1 bioRxiv preprint doi: https://doi.org/10.1101/177097; this version posted October 23, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Polycomb Group proteins are essential epigenetic repressors. They form multiple protein complexes of which two kinds, PRC1 and PRC2, are indispensable for repression. Although much is known about their biochemical properties, how PRC1 and PRC2 are targeted to specific genes is poorly understood. Here we establish the Cyclin D2 (CCND2) oncogene as a simple model to address this question. We provide the evidence that coordinated recruitment of PRC1 and PRC2 complexes to CCND2 involves a combination of a specialized PRC1 targeting element (PTE) and an adjacent CpG-island, which together act as a human Polycomb Response Element. -

Detection of H3k4me3 Identifies Neurohiv Signatures, Genomic
viruses Article Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis Liana Basova 1, Alexander Lindsey 1, Anne Marie McGovern 1, Ronald J. Ellis 2 and Maria Cecilia Garibaldi Marcondes 1,* 1 San Diego Biomedical Research Institute, San Diego, CA 92121, USA; [email protected] (L.B.); [email protected] (A.L.); [email protected] (A.M.M.) 2 Departments of Neurosciences and Psychiatry, University of California San Diego, San Diego, CA 92103, USA; [email protected] * Correspondence: [email protected] Abstract: Human postmortem specimens are extremely valuable resources for investigating trans- lational hypotheses. Tissue repositories collect clinically assessed specimens from people with and without HIV, including age, viral load, treatments, substance use patterns and cognitive functions. One challenge is the limited number of specimens suitable for transcriptional studies, mainly due to poor RNA quality resulting from long postmortem intervals. We hypothesized that epigenomic Citation: Basova, L.; Lindsey, A.; signatures would be more stable than RNA for assessing global changes associated with outcomes McGovern, A.M.; Ellis, R.J.; of interest. We found that H3K27Ac or RNA Polymerase (Pol) were not consistently detected by Marcondes, M.C.G. Detection of H3K4me3 Identifies NeuroHIV Chromatin Immunoprecipitation (ChIP), while the enhancer H3K4me3 histone modification was Signatures, Genomic Effects of abundant and stable up to the 72 h postmortem. We tested our ability to use H3K4me3 in human Methamphetamine and Addiction prefrontal cortex from HIV+ individuals meeting criteria for methamphetamine use disorder or not Pathways in Postmortem HIV+ Brain (Meth +/−) which exhibited poor RNA quality and were not suitable for transcriptional profiling. -
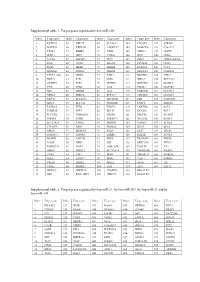
Target Genes Regulated by Hsa-Mir-21, by Hsa-Mir-203, by Hsa-Mir-21 and by Hsa-Mir-143
Supplemental table 1: Target genes regulated by hsa-miR-205 Index Target gene Index Target gene Index Target gene Index Target gene Index Target gene 1 KCTD20 35 UBE2Z 69 SLC38A1 103 LPCAT1 137 STK38L 2 MAPK14 36 YWHAH 70 ANGPTL7 104 MARCKS 138 C1orf123 3 TXNL1 37 RBBP4 71 CTGF 105 MED13 139 GUCD1 4 SPDL1 38 LRP1 72 CYR61 106 IPO7 140 CDK6 5 TCF20 39 IMPAD1 73 TP73 107 PHC2 141 CDKN2AIPNL 6 RAN 40 GNAS 74 EGLN2 108 PICALM 142 CLIP1 7 RGS6 41 MED1 75 ERBB2 109 PLAGL2 143 CUL5 8 HOXA11 42 INPPL1 76 PRRG4 110 NDUFA4 144 C6orf201 9 PAPPA-AS1 43 DDX5 77 F2RL2 111 NDUFB2 145 VTI1A 10 PRR15 44 E2F1 78 GOT1 112 NIPA2 146 SLC5A12 11 ACTRT3 45 E2F5 79 NUFIP2 113 NOTCH2 147 MAML2 12 YES1 46 ZEB2 80 IL24 114 PANK1 148 MAP3K9 13 SRC 47 ERBB3 81 IL32 115 PARD6B 149 NUDT21 14 NPRL3 48 PRKCE 82 RNF217 116 TMEM66 150 DNAJA1 15 NFAT5 49 SLC41A1 83 ZNF585B 117 EZR 151 CCDC108 16 XPOT 50 SLC7A2 84 SIGMAR1 118 ENPP4 152 SHISA6 17 KCTD16 51 ZEB1 85 VEGFA 119 LRRTM4 153 ACP1 18 TMSB4X 52 PHF8 86 BCL9L 120 KCNJ10 154 BCL2 19 PLCXD2 53 TMEM201 87 CREB1 121 PHLPP2 155 NCAPG 20 TNFSF8 54 PTPRJ 88 SERINC3 122 YEATS2 156 KLHL5 21 SLC25A25 55 ETNK1 89 HMGB3 123 VAMP1 157 ACSL4 22 C11orf74 56 XPR1 90 SRD5A1 124 RTN3 158 BCL6 23 GM2A 57 MRPL44 91 PTEN 125 RFX7 159 ITGA5 24 SMNDC1 58 TM9SF2 92 ESRRG 126 RAP2B 160 ACSL1 25 BAMBI 59 PAIP2B 93 PRLR 127 TRAF3IP1 161 EID2B 26 LCOR 60 NEK9 94 ICK 128 SERTAD2 162 TEX35 27 TMEM239 61 NOX5 95 LOH12CR1 129 TOLLIP 163 YY1 28 AMOT 62 DMXL2 96 SLC39A14 130 TMEM55B 164 SMAD1 29 CDK1 63 ETF1 97 BDP1 131 TMEM123 165 SMAD4 30 SQLE 64 -

Bmi1 – a Path to Targeting Cancer Stem Cells
Review Cancer Stem Cells Bmi1 – A Path to Targeting Cancer Stem Cells David Bakhshinyan,1,2 Ashley A Adile,1,2 Chitra Venugopal1,3 and Sheila K Singh1,2,3 1. McMaster Stem Cell and Cancer Research Institute, McMaster University, Hamilton, ON, Canada; 2. Department of Biochemistry and Biomedical Sciences, Faculty of Health Sciences, McMaster University, Hamilton, ON, Canada; 3. Department of Surgery, Faculty of Health Sciences, McMaster University, Hamilton, ON, Canada DOI: https://doi.org/10.17925/EOH.2017.13.02.147 he Polycomb group (PcG) genes encode for proteins comprising two multiprotein complexes, Polycomb repressive complex 1 (PRC1) and Polycomb repressive complex 2 (PRC2). Although the initial discovery of PcG genes was made in Drosophila, as transcriptional T repressors of homeotic (HOX) genes. Polycomb repressive complexes have been since implicated in regulating a wide range of cellular processes, including differentiation and self-renewal in normal and cancer stem cells. Bmi1, a subunit of PRC1, has been long implicated in driving self-renewal, the key property of stem cells. Subsequent studies showing upregulation of Bmi1 in several cancers correlated with increased aggressiveness, radioresistance and metastatic potential, provided rationale for development of targeted therapies against Bmi1. Although Bmi1 activity can be reduced through transcriptional, post-transcriptional and post-translational regulation, to date, the most promising approach has been through small molecule inhibitors targeting Bmi1 activity. The post-translational targeting of Bmi1 in colorectal carcinoma, lung adenocarcinoma, multiple myeloma and medulloblastoma have led to significant reduction of self-renewal capacity of cancer stem cells, leading to slower tumour progression and reduced extent of metastatic spread.