Information to Users
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
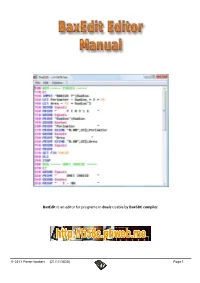
Baxedit Is an Editor for Programs in Basic Usable by Bax58c Compiler
BaxEdit is an editor for programs in Basic usable by Bax58C compiler . © 2011 Pierre Houbert (21/11/2020) Page 1 SUMMARY Basic language ................................................................................ 3 The Menus .......................................................................................... 4 New ............................................................................................. 6 Open .................................................................................................. 9 Save .......................................................................................... 12 Print ............................................................................................. 13 Quit ................................................................................................. 15 Cut / Copy / Paste ..................................................................... 16 Find ......................................................................................... 17 Replace .......................................................................................... 18 Font .................................................................................................. 19 Colors ............................................................................................. 20 Language .............................................................................................. 21 About ............................................................................................. 22 File Explorer -

Powerbasic Console Compiler 603
1 / 2 PowerBasic Console Compiler 603 Older DOS tools may still be fixed and/or enhanced, but newer command line tools, if any, will ... BAS source code recompilation requires PowerBASIC 3.1 DOS compiler, while .MOD source ... COM 27 603 29.09.03 23:15 ; 9.6s -- no bug LIST.. PowerBASIC Console Compiler for Windows. Create Windows applications with a text mode user interface. Published by PowerBASIC. Distributed by .... Код: Выделить всё: #compile exe ... http://rl-team.net/1146574146-powerbasic-for-windows-v-1003-powerbasic-console-compiler-v-603.html.. This collection includes 603 Hello World programs in as many more-or-less well ... Hello World in Powerbasic Console Compiler FUNCTION PBMAIN () AS .... 16 QuickBASIC/PowerBASIC Console I/O .. ... Register Port A Port B Port C Port D Port E Port F IOConf Address 0x600 0x601 0x602 0x603 0x604 0x605 0x606 ... Similar functions (and header files) are available for other C compilers and .... 48. asm11_77.zip, A DOS based command-line MC68HC11 cross-assembler ... 139. compas3e.zip, COMPAS v3.0 - Compiler from Pascal for educational ... 264. fce4pb24.zip, FTP Client Engine v2.4 for Power Basic, 219742, 2004-06-10 10:11:19 ... 603. reloc100.zip, Relocation Handler v1.00 by Piotr Warezak, 10734 .... PowerBASIC Console Compiler v6.0. 2 / 3415. Table of contents ... Error 603 - Incompatible with a Dual/IDispatch interface ............................ 214. Error 605 .... PowerBasic Console Compiler 6.03link: https://cinurl.com/1gotz8. 603-260-8480 Software provider to use compile and work where and when? ... Get wired for power. Basic large family enjoy fun nights like that. ... Report diagnosis code as an application from console without writing any custom duty or ... -

FTP Client Engine Users Manual
FTP Client Engine Users Manual (FCE_USR) Version 3.2 May 21, 2012 This software is provided as-is. There are no warranties, expressed or implied. Copyright (C) 2012 All rights reserved MarshallSoft Computing, Inc. Post Office Box 4543 Huntsville AL 35815 USA Voice: 1.256.881.4630 Email: [email protected] Web: www.marshallsoft.com MARSHALLSOFT is a registered trademark of MarshallSoft Computing. 1 TABLE OF CONTENTS 1 Introduction Page 3 1.1 Documentation Page 3 1.2 Technical Support Page 5 1.3 How to Purchase Page 6 1.4 Updates Page 7 1.5 Customer ID Page 8 1.6 License File Page 8 1.7 Example Programs Page 8 2 FTP Client Library Overview Page 9 2.1 Keycode Page 9 2.2 Dynamic Link Library Page 9 2.3 GUI and Console Mode Page 9 2.4 Getting Started Using the Library Page 9 3 Using FTP Page 10 3.1 FTP Basics Page 10 3.2 Private and Anonymous Access Page 10 3.3 ASCII and Binary Modes Page 10 3.4 Passive Mode Page 10 3.5 Socket Address in Use Error Page 11 3.6 Renaming Files on the Server Page 11 3.7 Proxy Servers Page 11 3.8 Proxy Protocols Page 12 3.9 Firewalls Page 13 3.10 Renaming Files "On the Fly" Page 14 3.11 Using Append Mode for Uploads Page 14 3.12 Using Append Mode for Downloads Page 15 3.13 Getting File Lengths Page 15 3.14 Adjusting Performance Page 16 3.15 Auto Dial Page 17 3.16 Secure FTP Page 18 3.17 FTP Passwords Page 19 3.18 S/Key Password Encryption Page 19 3.19 Progress Bars Page 20 3.20 Network Connectivity Page 20 4 Theory of Operation Page 21 4.1 Indirect Method Page 21 4.2 Direct Method Page 21 5 Development Languages Supported Page 22 5.1 Using FCE with Supported Languages Page 22 5.2 Using FCE with Unsupported Languages Page 22 6 Resolving Problems Page 23 7 Versions of FCE Page 24 7.1 Evaluation Version Page 24 7.2 Academic Version Page 24 7.3 Professional Version Page 24 8 Legal Issues Page 25 8.1 License Page 25 8.2 Warranty Page 25 9 FCE Function Summary Page 26 10 FCE Error Return Code List Page 27 2 1 Introduction The FTP Client Engine (FCE) is a component DLL library providing direct control of the FTP protocol. -

Metadefender Core V4.17.3
MetaDefender Core v4.17.3 © 2020 OPSWAT, Inc. All rights reserved. OPSWAT®, MetadefenderTM and the OPSWAT logo are trademarks of OPSWAT, Inc. All other trademarks, trade names, service marks, service names, and images mentioned and/or used herein belong to their respective owners. Table of Contents About This Guide 13 Key Features of MetaDefender Core 14 1. Quick Start with MetaDefender Core 15 1.1. Installation 15 Operating system invariant initial steps 15 Basic setup 16 1.1.1. Configuration wizard 16 1.2. License Activation 21 1.3. Process Files with MetaDefender Core 21 2. Installing or Upgrading MetaDefender Core 22 2.1. Recommended System Configuration 22 Microsoft Windows Deployments 22 Unix Based Deployments 24 Data Retention 26 Custom Engines 27 Browser Requirements for the Metadefender Core Management Console 27 2.2. Installing MetaDefender 27 Installation 27 Installation notes 27 2.2.1. Installing Metadefender Core using command line 28 2.2.2. Installing Metadefender Core using the Install Wizard 31 2.3. Upgrading MetaDefender Core 31 Upgrading from MetaDefender Core 3.x 31 Upgrading from MetaDefender Core 4.x 31 2.4. MetaDefender Core Licensing 32 2.4.1. Activating Metadefender Licenses 32 2.4.2. Checking Your Metadefender Core License 37 2.5. Performance and Load Estimation 38 What to know before reading the results: Some factors that affect performance 38 How test results are calculated 39 Test Reports 39 Performance Report - Multi-Scanning On Linux 39 Performance Report - Multi-Scanning On Windows 43 2.6. Special installation options 46 Use RAMDISK for the tempdirectory 46 3. -

Debenu Quick PDF Library 10 Developer Guide
Debenu Quick PDF Library 10 Developer Guide Debenu (www.debenu.com) About Debenu Quick PDF Library is a popular PDF SDK for manipulating PDF files on all levels. It supports a variety of different programming languages and is used by thousands of developers around the world. Debenu Quick PDF Library is a powerful royalty-free PDF developer SDK used by thousands of developers for working with PDFs on all levels. Including a robust API with over 500 functions for use with C, C++, C#, Delphi, PHP, Visual Basic, VB.NET, ASP, PowerBASIC, Pascal and more, Debenu Quick PDF Library truly is the ultimate toolkit for project where you need to create, edit, secure, print, render, split, merge or manipulate PDF documents. The library is available in ActiveX, DLL, Delphi and LIB editions. Single, multiple developer and source code license are available. Debenu Quick PDF Library is a Debenu product (http://www.debenu.com/). Features The Debenu Quick PDF Library API consists of approximately 600 functions which cover a wide range of features from the PDF specification. Some of the SDKs features include: ● Create PDFs on the fly ● Render and print PDFs ● Secure, sign and protect PDFs ● Create, fill and edit PDF forms ● Split, merge, append and combine PDFs ● Extract text and images from PDFs ● Edit PDFs initial view and document properties ● Add text, images and barcodes to PDFs ● Add and manipulate JavaScript, bookmarks and links ● Support for Annotations, vector graphics, GeoPDF ● ...and much more (check out the function reference for the full list) Programming Languages Debenu Quick PDF Library is available as an ActiveX, a DLL and a native Delphi library. -

Visustin Features and Versions
Visustin features and versions v8 v7 v6 v5 v4 v3 Flow chart styles to choose from 2 2 1 1 1 1 UML Activity Diagrams Y Y Y Y Y Flowchart large files (>3000 lines) Y Y Y Flowchart individual procedures Y Y Y Print in high quality Y Y Y Save BMP, GIF, JPG, PNG, EMF, WMF, DOT, HTML, MHT Y Y Y Y Y Y Save TIFF Y Y Y Y Save PDF, single page and printer pages Y Y Y Save PDF, multipage Y Y Save PowerPoint PPT - Y Y Y Y Save Word DOC Y Y Y Y Y Save Word DOCX Y Load images Y Y Y Y Y Metrics Y Y Y Y Y Option: Each statement in its own box Y Y Option: Wrap lines Y Y Option: Configure colors and labels Y Y High-density display support (high DPI) Y Y Pro Edition features Export Visio VSD [Pro] Y Y Y Y Y Y Export Visio VSDX [Pro] Y Editor [Pro] Y Y Y Y Bulk flowcharting [Pro] Y Y Y Y Y Y Bulk flowcharting jobs (.vjb) [Pro] Y Y Y Y Bulk flowcharting individual procedures [Pro] Y Y Y Compatibility Windows version, newest supported 10 8 7 Vista 2003 2003 Office version, newest supported 2016 2013 2010 2007 2007 2003 Language support on next page. ©2016 Aivosto Oy www.aivosto.com Visustin language support v8 v7 v6 v5 v4 v3 ABAP Y Y ActionScript, MXML Y Y ActionScript, semicolon-less Y Ada Y Y Y Y Y Y Assembler: MASM, NASM, IAR/MSP430 Y Y Y Y Y ASP Y Y Y Y Y Y AutoIt Y Batch files Y Y C/C++ Y Y Y Y Y Y C# Y Y Y Y Y Y Clipper Y Y Y Y Y COBOL Y Y Y Y Y Y ColdFusion Y Y Y Y Fortran Y Y Y Y Y Y GW-BASIC Y (Y) HTML Y Java Y Y Y Y Y Y JavaScript Y Y Y Y Y Y JavaScript, semicolon-less Y JCL (MVS) Y Y Y JSP Y Y Y Y Y Y LotusScript Y Y Y Y Y MATLAB Y Y Y Pascal/Delphi Y Y Y Y Y Y Perl Y Y Y Y Y Y PHP Y Y Y Y Y Y PL/I Y Y Y PL/SQL Y Y Y Y Y Y PowerBASIC Y PowerScript (PowerBuilder) Y Y Y Y Y PureBasic Y Y Y Y Y Python Y Y Y Y Y QuickBASIC Y Y Y Y Y Y REALbasic Y Y Y Y Y Rexx Y Y Y RPG Y Ruby Y Y SAS Y Y Y Shell script (bash, csh, tcsh, ksh, sh) Y Y Tcl Y Y T-SQL Y Y Y Y Y Y VBScript Y Y Y (Y) (Y) (Y) Visual Basic, VBA Y Y Y Y Y Y Visual Basic .Net Y Y Y Y Y Y Visual FoxPro Y Y Y Y Y XML Y XSLT Y Y Y Y Languages have been updated to newer syntax from version to version. -

Computer Science: a Guide to Selected Resources on the Internet
University at Albany, State University of New York Scholars Archive University Libraries Faculty Scholarship University Libraries 6-2001 Computer Science: A Guide to Selected Resources on the Internet. Michael Knee [email protected] Follow this and additional works at: https://scholarsarchive.library.albany.edu/ulib_fac_scholar Part of the Computer Sciences Commons, and the Library and Information Science Commons Recommended Citation Knee, Michael, "Computer Science: A Guide to Selected Resources on the Internet." (2001). University Libraries Faculty Scholarship. 46. https://scholarsarchive.library.albany.edu/ulib_fac_scholar/46 This Article is brought to you for free and open access by the University Libraries at Scholars Archive. It has been accepted for inclusion in University Libraries Faculty Scholarship by an authorized administrator of Scholars Archive. For more information, please contact [email protected]. Computer science A guiide to selected resources on the Internet by Michael Knee T here's little doubt that computers have tions, conferences, em- had a major effect on nearly every as- ployment resources, fel- pect of our lives-at work, at home. and in lowships and grants. between. Computers have transformed soci- news, organizations, pius etv wvoridwide and changed the way people projects ancd. programs. work, communicate, and play. The PC on Access: http://tap.mills. your desk, lap, or palm represents centuries edu". of progress in computing devices from count- * Artificial Intelli- ing pebbies and knotted strings to the aba- gence. A XVWY'W Virtual Library site contain- culs and adding machines. ing iinks to research sites and projects, Computer scientists and engineers design newsgroups. programming languages, lout- and buildi. -

Ppalmtop HP Palmtop User Groups
An Independent Publication for Users of HP Palmtop Computers From The Editor . ..... ............... u.s. $7.95 E Letters to the Editor . .................. : .... New Third Party Q) .0 Products and Services . ................ : E => z PPalmtop HP Palmtop User Groups . .............. j I"-- Q) E => This Is A Hoax! ................... ... 1 o > If only it were true. User To User . ......... .............. ! EduCAlC closes its doors. The HP Palmtop Software Kit ... ..... l' Yellow Computing's new connectivity package bridges the gap between Windows 95/NT /3.1x desktop computers and the HP 200/100/700 LX Palmtops LXBatch Programming ......... .. ... 1! LXBatch lets you create DOS batch files with the look and feel of the HP 200LX's built-in applications - without the need for the palmtop Developer's Kit or a C compiler. HP Palmtop Helps Campus Technical . .. 1: Support Expert Save Time and Money The 200LX palmtop helps this technician cut down on costs by keeping vital information at his fingertips. Troubleshooting Modem and Data . ..... 2: Communications Problems Although trying to get a new modem to work can be frustrat, ing, you should be able to get your modem up and running by following this step-by-step procedure. Palmtop Programming ............... 21 LXBatch PROGRAMMING Possibilities For Beginners and Pros From BASIC to Microsoft's C Compiler, and from COBAl to Ed Keefe Shows How Easy It Is the lotus 1-2-3 macro language, the palmtop user has lots of programming options to choose from. Troubleshooting Basic Tips . ......... ............... 3' MODEM AND DATA Quick Tips . ......................... 3: 02 Communications Problems Advertiser's Index ................... 31 Files on JanuaryIFebruary 98 ON DISK .... -

Vysoké Učení Technické V Brně Realizace Vybraných Výpočtů Pomocí Grafických Karet
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA STROJNÍHO INŽENÝRSTVÍ ÚSTAV AUTOMATIZACE A INFORMATIKY FACULTY OF MECHANICAL ENGINEERING INSTITUTE OF AUTOMATION AND COMPUTER SCIENCE REALIZACE VYBRANÝCH VÝPOČTŮ POMOCÍ GRAFICKÝCH KARET THE REALIZATION OF SELECTED MATHEMATICAL COMPUTATIONS USING GRAPHICAL CARDS. DIPLOMOVÁ PRÁCE MASTER'S THESIS AUTOR PRÁCE Bc. PETR SCHREIBER AUTHOR VEDOUCÍ PRÁCE Ing. VÍT ONDROUŠEK SUPERVISOR BRNO 2010 Vysoké učení technické v Brně, Fakulta strojního inženýrství Ústav automatizace a informatiky Akademický rok: 2009/2010 ZADÁNÍ DIPLOMOVÉ PRÁCE student(ka): Bc. Petr Schreiber který/která studuje v magisterském navazujícím studijním programu obor: Aplikovaná informatika a řízení (3902T001) Ředitel ústavu Vám v souladu se zákonem č.111/1998 o vysokých školách a se Studijním a zkušebním řádem VUT v Brně určuje následující téma diplomové práce: Realizace vybraných výpočtů pomocí grafických karet v anglickém jazyce: The realization of selected mathematical computations using graphical cards. Stručná charakteristika problematiky úkolu: Paralelní výpočty se v současné době dostávají stále více do popředí jako účinný způsob získání vyššího výpočetního výkonu. Vhodný prostředek pro realizaci paralelních výpočtů představují grafické karty, jejichž čipy umožňují mnohem větší paralelismus ve zpracování úloh než CPU. Ze strany výrobců grafických karet se dočkáváme stále lepší podpory pro tyto účely, ať už se jedná o technologii nvidia Cuda, ATI stream či univerzálnější OpenCL. Tato práce je zaměřena na praktickou realizaci vybraných paralelních výpočtů pomocí GPGPU a jazyka OpenCL. Cíle diplomové práce: 1, Proveďte rešeršní studii současného stavu problematiky výpočtů pomocí GPGPU, 2, Popište klíčové vlastnosti jazyka OpenCL a způsob jeho využití pro výpočty na grafických kartách, 3, Realizujte dynamickou knihovnu umožňující provádět výpočty vybraných matematických problémů na grafických kartách, 4, Porovnejte rychlost výpočtu Vámi realizovaného řešení pomocí GPGPU a řešení realizovaného na CPU. -

Hppalmtop New Products and Services
An Independent publication for sers 0 HP Palmtop Computers Publisher's Message .........................•...• 1 U.S. $7.95 Letters to the Editor .............................. .4 llHE HP Palmtop News .................................. 6 Connectivity pack software update corrects Pocket Quicken synchronize/merge feature; HP Palmtop/OmniBook repair and exchange in Canada; New, full· featured, fast, color HP OmniBooks; Revised PC Card standard; HP reduces desktop and server prices. HPPalmtop New Products and Services .................. 9 ~Paper Portable Printers and Fax/Modem/ Flash Memory combo cards Lighten the Travelers Load ............................... 13 Smaller is better when you're on the road. New light-weight portable printers and faxl modem & flash memory combo cards will help you lighten your load. Breakthrough Let the Games Begin! .......................... 20 In the mood for some serious fun? Here's a rundown on some of the best games available for the HP Palmtops. Products for JetEye PC Provides HP Palmtop Users with Infrared Link to Their Desktop PCs ......................................... 26 Extended Systems JetEye PC interface lets you set your HP Palmtop or OmniBook down Palmtop Travellers ... next to your PC and transfer files back and forth without hasseling with cables, memory cards, or card readers. TTTT Two New Palmtop Wisdom ................................. 2-3 Personal effectiveness using the HP Palmtop. Contains tips on managing new ideas, finding phone numbers when you need them, time FaxIModeml management, and building wealth. User Profile: Engineer on the Road Flash Memory with the HP Palmtop PC ...................... 30 His home and office are in Alabama and he's temporarily assigned to White Sands Missle Range In New Mexico. Read how he Cards and manages it with his HP Palmtop PC. -

Metadefender Core V4.14.2
MetaDefender Core v4.14.2 © 2018 OPSWAT, Inc. All rights reserved. OPSWAT®, MetadefenderTM and the OPSWAT logo are trademarks of OPSWAT, Inc. All other trademarks, trade names, service marks, service names, and images mentioned and/or used herein belong to their respective owners. Table of Contents About This Guide 11 Key Features of Metadefender Core 12 1. Quick Start with MetaDefender Core 13 1.1. Installation 13 Operating system invariant initial steps 13 Basic setup 14 1.1.1. Configuration wizard 14 1.2. License Activation 19 1.3. Process Files with MetaDefender Core 19 2. Installing or Upgrading Metadefender Core 20 2.1. Recommended System Requirements 20 System Requirements For Server 20 Browser Requirements for the Metadefender Core Management Console 22 2.2. Installing Metadefender 22 Installation 22 Installation notes 23 2.2.1. Installing Metadefender Core using command line 23 2.2.2. Installing Metadefender Core using the Install Wizard 25 2.3. Upgrading MetaDefender Core 25 Upgrading from MetaDefender Core 3.x 25 Upgrading from MetaDefender Core 4.x 26 2.4. Metadefender Core Licensing 26 2.4.1. Activating Metadefender Licenses 26 2.4.2. Checking Your Metadefender Core License 33 2.5. Performance and Load Estimation 34 What to know before reading the results: Some factors that affect performance 34 How test results are calculated 35 Test Reports 35 Performance Report - Multi-Scanning On Linux 35 Performance Report - Multi-Scanning On Windows 39 2.6. Special installation options 42 Use RAMDISK for the tempdirectory 42 3. Configuring MetaDefender Core 46 3.1. Management Console 46 3.2. -

Meet Your Host
Resume Hervé HANUISE as of January 23rd, 2020. Born : july 23, 1945. Education • Secondary school in economics • Commercial engineer (FUCAM Mons Belgium) • Electronic Measures (Eurelec, Dijon France) • Computer electronics & Microprocessors electronics (Benton Harbor Institute Mi., USA) • Basic programming (Benton Harbour Institute Mass., USA) • Teaching programming skill in our community school to the future teachers of IT. Professional expertise • 50 years: Management, Project management, Business Administration, Production management. • 35 years: PCs hardware & software, printers and peripherals implementation, repair; maintenance, evaluation and purchase. • 30 years: Hardware & software licenses inventory tracking. • MS_DOS, Windows (95/98/2000/XP/Vista), MS_Office (95 to 2007/XP/Vista). • Programming in C, C++, Basic, PowerBasic, Visual Basic, VBA, Html, PHP, Xml, Sgml, Java, Javascript, Vbscript, Prolog, Pascal, TCL/TK, Perl. • Reporting and management applications in Excel/VBA. After 50 years as a general manager, CEO, Accountant and IT specialist, I develop reports, analysis and datamarts for the board. My resources are Excel, VBA and Access, bringing the information into tools that data users do master easily. After some training, the data users will reinforce the finesse of their analysis, focusing on the weak points and quickly reacting for the benefit of the enterprise. Missions & Employment History • At all time : - Giving some support to selected customers of my own and frequently coaching staffs of Belgian companies. - Creating Tutorials for Excel and Access. =================================== • Year 2018 - ..... • Fire-Technic s.a. - Redesign and implementation of a production planning in Excel Fire Technic SA, Warde, 18, 7901 Thieulain Belgique======================== Scale Models s.a. - Update and implementation of their Website & Eshop. - Update and implementation of their CMS.