Electron-Nucleus Interactions and Its Biophysical Consequences
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
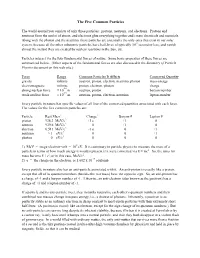
The Five Common Particles
The Five Common Particles The world around you consists of only three particles: protons, neutrons, and electrons. Protons and neutrons form the nuclei of atoms, and electrons glue everything together and create chemicals and materials. Along with the photon and the neutrino, these particles are essentially the only ones that exist in our solar system, because all the other subatomic particles have half-lives of typically 10-9 second or less, and vanish almost the instant they are created by nuclear reactions in the Sun, etc. Particles interact via the four fundamental forces of nature. Some basic properties of these forces are summarized below. (Other aspects of the fundamental forces are also discussed in the Summary of Particle Physics document on this web site.) Force Range Common Particles It Affects Conserved Quantity gravity infinite neutron, proton, electron, neutrino, photon mass-energy electromagnetic infinite proton, electron, photon charge -14 strong nuclear force ≈ 10 m neutron, proton baryon number -15 weak nuclear force ≈ 10 m neutron, proton, electron, neutrino lepton number Every particle in nature has specific values of all four of the conserved quantities associated with each force. The values for the five common particles are: Particle Rest Mass1 Charge2 Baryon # Lepton # proton 938.3 MeV/c2 +1 e +1 0 neutron 939.6 MeV/c2 0 +1 0 electron 0.511 MeV/c2 -1 e 0 +1 neutrino ≈ 1 eV/c2 0 0 +1 photon 0 eV/c2 0 0 0 1) MeV = mega-electron-volt = 106 eV. It is customary in particle physics to measure the mass of a particle in terms of how much energy it would represent if it were converted via E = mc2. -

A Discussion on Characteristics of the Quantum Vacuum
A Discussion on Characteristics of the Quantum Vacuum Harold \Sonny" White∗ NASA/Johnson Space Center, 2101 NASA Pkwy M/C EP411, Houston, TX (Dated: September 17, 2015) This paper will begin by considering the quantum vacuum at the cosmological scale to show that the gravitational coupling constant may be viewed as an emergent phenomenon, or rather a long wavelength consequence of the quantum vacuum. This cosmological viewpoint will be reconsidered on a microscopic scale in the presence of concentrations of \ordinary" matter to determine the impact on the energy state of the quantum vacuum. The derived relationship will be used to predict a radius of the hydrogen atom which will be compared to the Bohr radius for validation. The ramifications of this equation will be explored in the context of the predicted electron mass, the electrostatic force, and the energy density of the electric field around the hydrogen nucleus. It will finally be shown that this perturbed energy state of the quan- tum vacuum can be successfully modeled as a virtual electron-positron plasma, or the Dirac vacuum. PACS numbers: 95.30.Sf, 04.60.Bc, 95.30.Qd, 95.30.Cq, 95.36.+x I. BACKGROUND ON STANDARD MODEL OF COSMOLOGY Prior to developing the central theme of the paper, it will be useful to present the reader with an executive summary of the characteristics and mathematical relationships central to what is now commonly referred to as the standard model of Big Bang cosmology, the Friedmann-Lema^ıtre-Robertson-Walker metric. The Friedmann equations are analytic solutions of the Einstein field equations using the FLRW metric, and Equation(s) (1) show some commonly used forms that include the cosmological constant[1], Λ. -

Lesson 1: the Single Electron Atom: Hydrogen
Lesson 1: The Single Electron Atom: Hydrogen Irene K. Metz, Joseph W. Bennett, and Sara E. Mason (Dated: July 24, 2018) Learning Objectives: 1. Utilize quantum numbers and atomic radii information to create input files and run a single-electron calculation. 2. Learn how to read the log and report files to obtain atomic orbital information. 3. Plot the all-electron wavefunction to determine where the electron is likely to be posi- tioned relative to the nucleus. Before doing this exercise, be sure to read through the Ins and Outs of Operation document. So, what do we need to build an atom? Protons, neutrons, and electrons of course! But the mass of a proton is 1800 times greater than that of an electron. Therefore, based on de Broglie’s wave equation, the wavelength of an electron is larger when compared to that of a proton. In other words, the wave-like properties of an electron are important whereas we think of protons and neutrons as particle-like. The separation of the electron from the nucleus is called the Born-Oppenheimer approximation. So now we need the wave-like description of the Hydrogen electron. Hydrogen is the simplest atom on the periodic table and the most abundant element in the universe, and therefore the perfect starting point for atomic orbitals and energies. The compu- tational tool we are going to use is called OPIUM (silly name, right?). Before we get started, we should know what’s needed to create an input file, which OPIUM calls a parameter files. Each parameter file consist of a sequence of ”keyblocks”, containing sets of related parameters. -

1.1. Introduction the Phenomenon of Positron Annihilation Spectroscopy
PRINCIPLES OF POSITRON ANNIHILATION Chapter-1 __________________________________________________________________________________________ 1.1. Introduction The phenomenon of positron annihilation spectroscopy (PAS) has been utilized as nuclear method to probe a variety of material properties as well as to research problems in solid state physics. The field of solid state investigation with positrons started in the early fifties, when it was recognized that information could be obtained about the properties of solids by studying the annihilation of a positron and an electron as given by Dumond et al. [1] and Bendetti and Roichings [2]. In particular, the discovery of the interaction of positrons with defects in crystal solids by Mckenize et al. [3] has given a strong impetus to a further elaboration of the PAS. Currently, PAS is amongst the best nuclear methods, and its most recent developments are documented in the proceedings of the latest positron annihilation conferences [4-8]. PAS is successfully applied for the investigation of electron characteristics and defect structures present in materials, magnetic structures of solids, plastic deformation at low and high temperature, and phase transformations in alloys, semiconductors, polymers, porous material, etc. Its applications extend from advanced problems of solid state physics and materials science to industrial use. It is also widely used in chemistry, biology, and medicine (e.g. locating tumors). As the process of measurement does not mostly influence the properties of the investigated sample, PAS is a non-destructive testing approach that allows the subsequent study of a sample by other methods. As experimental equipment for many applications, PAS is commercially produced and is relatively cheap, thus, increasingly more research laboratories are using PAS for basic research, diagnostics of machine parts working in hard conditions, and for characterization of high-tech materials. -

A Young Physicist's Guide to the Higgs Boson
A Young Physicist’s Guide to the Higgs Boson Tel Aviv University Future Scientists – CERN Tour Presented by Stephen Sekula Associate Professor of Experimental Particle Physics SMU, Dallas, TX Programme ● You have a problem in your theory: (why do you need the Higgs Particle?) ● How to Make a Higgs Particle (One-at-a-Time) ● How to See a Higgs Particle (Without fooling yourself too much) ● A View from the Shadows: What are the New Questions? (An Epilogue) Stephen J. Sekula - SMU 2/44 You Have a Problem in Your Theory Credit for the ideas/example in this section goes to Prof. Daniel Stolarski (Carleton University) The Usual Explanation Usual Statement: “You need the Higgs Particle to explain mass.” 2 F=ma F=G m1 m2 /r Most of the mass of matter lies in the nucleus of the atom, and most of the mass of the nucleus arises from “binding energy” - the strength of the force that holds particles together to form nuclei imparts mass-energy to the nucleus (ala E = mc2). Corrected Statement: “You need the Higgs Particle to explain fundamental mass.” (e.g. the electron’s mass) E2=m2 c4+ p2 c2→( p=0)→ E=mc2 Stephen J. Sekula - SMU 4/44 Yes, the Higgs is important for mass, but let’s try this... ● No doubt, the Higgs particle plays a role in fundamental mass (I will come back to this point) ● But, as students who’ve been exposed to introductory physics (mechanics, electricity and magnetism) and some modern physics topics (quantum mechanics and special relativity) you are more familiar with.. -

1 Solving Schrödinger Equation for Three-Electron Quantum Systems
Solving Schrödinger Equation for Three-Electron Quantum Systems by the Use of The Hyperspherical Function Method Lia Leon Margolin 1 , Shalva Tsiklauri 2 1 Marymount Manhattan College, New York, NY 2 New York City College of Technology, CUNY, Brooklyn, NY, Abstract A new model-independent approach for the description of three electron quantum dots in two dimensional space is developed. The Schrödinger equation for three electrons interacting by the logarithmic potential is solved by the use of the Hyperspherical Function Method (HFM). Wave functions are expanded in a complete set of three body hyperspherical functions. The center of mass of the system and relative motion of electrons are separated. Good convergence for the ground state energy in the number of included harmonics is obtained. 1. Introduction One of the outstanding achievements of nanotechnology is construction of artificial atoms- a few-electron quantum dots in semiconductor materials. Quantum dots [1], artificial electron systems realizable in modern semiconductor structures, are ideal physical objects for studying effects of electron-electron correlations. Quantum dots may contain a few two-dimensional (2D) electrons moving in the plane z=0 in a lateral confinement potential V(x, y). Detailed theoretical study of physical properties of quantum-dot atoms, including the Fermi-liquid-Wigner molecule crossover in the ground state with growing strength of intra-dot Coulomb interaction attracted increasing interest [2-4]. Three and four electron dots have been studied by different variational methods [4-6] and some important results for the energy of states have been reported. In [7] quantum-dot Beryllium (N=4) as four Coulomb-interacting two dimensional electrons in a parabolic confinement was investigated. -

One-Electron Atom
One-electron Atom The atomic orbitals of hydrogen-like atoms are solutions to the Schrödinger equation in a spherically symmetric potential. In this case, the potential term is the potential given by Coulomb's law: where •ε0 is the permittivity of the vacuum, •Z is the atomic number (number of protons in the nucleus), •e is the elementary charge (charge of an electron), •r is the distance of the electron from the nucleus. After writing the wave function as a product of functions: (in spherical coordinates), where Ylm are spherical harmonics, we arrive at the following Schrödinger equation: 2010年10月11日星期一 where µ is, approximately, the mass of the electron. More accurately, it is the reduced mass of the system consisting of the electron and the nucleus. Different values of l give solutions with different angular momentum, where l (a non-negative integer) is the quantum number of the orbital angular momentum. The magnetic quantum number m (satisfying ) is the (quantized) projection of the orbital angular momentum on the z-axis. 2010年10月11日星期一 Wave function In addition to l and m, a third integer n > 0, emerges from the boundary conditions placed on R. The functions R and Y that solve the equations above depend on the values of these integers, called quantum numbers. It is customary to subscript the wave functions with the values of the quantum numbers they depend on. The final expression for the normalized wave function is: where: • are the generalized Laguerre polynomials in the definition given here. • Here, µ is the reduced mass of the nucleus-electron system, where mN is the mass of the nucleus. -

1 the Principle of Wave–Particle Duality: an Overview
3 1 The Principle of Wave–Particle Duality: An Overview 1.1 Introduction In the year 1900, physics entered a period of deep crisis as a number of peculiar phenomena, for which no classical explanation was possible, began to appear one after the other, starting with the famous problem of blackbody radiation. By 1923, when the “dust had settled,” it became apparent that these peculiarities had a common explanation. They revealed a novel fundamental principle of nature that wascompletelyatoddswiththeframeworkofclassicalphysics:thecelebrated principle of wave–particle duality, which can be phrased as follows. The principle of wave–particle duality: All physical entities have a dual character; they are waves and particles at the same time. Everything we used to regard as being exclusively a wave has, at the same time, a corpuscular character, while everything we thought of as strictly a particle behaves also as a wave. The relations between these two classically irreconcilable points of view—particle versus wave—are , h, E = hf p = (1.1) or, equivalently, E h f = ,= . (1.2) h p In expressions (1.1) we start off with what we traditionally considered to be solely a wave—an electromagnetic (EM) wave, for example—and we associate its wave characteristics f and (frequency and wavelength) with the corpuscular charac- teristics E and p (energy and momentum) of the corresponding particle. Conversely, in expressions (1.2), we begin with what we once regarded as purely a particle—say, an electron—and we associate its corpuscular characteristics E and p with the wave characteristics f and of the corresponding wave. -

Photon–Photon and Electron–Photon Colliders with Energies Below a Tev Mayda M
Photon–Photon and Electron–Photon Colliders with Energies Below a TeV Mayda M. Velasco∗ and Michael Schmitt Northwestern University, Evanston, Illinois 60201, USA Gabriela Barenboim and Heather E. Logan Fermilab, PO Box 500, Batavia, IL 60510-0500, USA David Atwood Dept. of Physics and Astronomy, Iowa State University, Ames, Iowa 50011, USA Stephen Godfrey and Pat Kalyniak Ottawa-Carleton Institute for Physics Department of Physics, Carleton University, Ottawa, Canada K1S 5B6 Michael A. Doncheski Department of Physics, Pennsylvania State University, Mont Alto, PA 17237 USA Helmut Burkhardt, Albert de Roeck, John Ellis, Daniel Schulte, and Frank Zimmermann CERN, CH-1211 Geneva 23, Switzerland John F. Gunion Davis Institute for High Energy Physics, University of California, Davis, CA 95616, USA David M. Asner, Jeff B. Gronberg,† Tony S. Hill, and Karl Van Bibber Lawrence Livermore National Laboratory, Livermore, CA 94550, USA JoAnne L. Hewett, Frank J. Petriello, and Thomas Rizzo Stanford Linear Accelerator Center, Stanford University, Stanford, California 94309 USA We investigate the potential for detecting and studying Higgs bosons in γγ and eγ collisions at future linear colliders with energies below a TeV. Our study incorporates realistic γγ spectra based on available laser technology, and NLC and CLIC acceleration techniques. Results include detector simulations.√ We study the cases of: a) a SM-like Higgs boson based on a devoted low energy machine with see ≤ 200 GeV; b) the heavy MSSM Higgs bosons; and c) charged Higgs bosons in eγ collisions. 1. Introduction The option of pursuing frontier physics with real photon beams is often overlooked, despite many interesting and informative studies [1]. -

Neutrino-Electron Scattering: General Constraints on Z 0 and Dark Photon Models
Neutrino-Electron Scattering: General Constraints on Z 0 and Dark Photon Models Manfred Lindner,a Farinaldo S. Queiroz,b Werner Rodejohann,a Xun-Jie Xua aMax-Planck-Institut für Kernphysik, Saupfercheckweg 1, 69117 Heidelberg, Germany bInternational Institute of Physics, Federal University of Rio Grande do Norte, Campus Univer- sitário, Lagoa Nova, Natal-RN 59078-970, Brazil E-mail: [email protected], [email protected], [email protected], [email protected] Abstract. We study the framework of U(1)X models with kinetic mixing and/or mass mixing terms. We give general and exact analytic formulas and derive limits on a variety of U(1)X models that induce new physics contributions to neutrino-electron scattering, taking into account interference between the new physics and Standard Model contributions. Data from TEXONO, CHARM-II and GEMMA are analyzed and shown to be complementary to each other to provide the most restrictive bounds on masses of the new vector bosons. In particular, we demonstrate the validity of our results to dark photon-like as well as light Z0 models. arXiv:1803.00060v1 [hep-ph] 28 Feb 2018 Contents 1 Introduction 1 2 General U(1)X Models2 3 Neutrino-Electron Scattering in U(1)X Models7 4 Data Fitting 10 5 Bounds 13 6 Conclusion 16 A Gauge Boson Mass Generation 16 B Cross Sections of Neutrino-Electron Scattering 18 C Partial Cross Section 23 1 Introduction The Standard Model provides an elegant and successful explanation to the electroweak and strong interactions in nature [1]. -

Physics Results from the First Electron-Proton Collider HERA
DESY <>r> -02r, March 1995 KS001829947 R: FI 0EC07443217 ■*DE007443217* Physics Results from the First Electron-Proton Collider HERA A. De Roeck Drutachcs Elektrotten-Synchrotron DESY. Hamburg DISTRIBUTION OF THIS DOCUMENT IS UNLIMITED FOREIGN SALES PROHIBITED ISSN 04IS-9S13 BEST 95-025 ISS35 0418-9833 March 1995 PHYSICS RESULTS FROM THE FIRST ELECTRON-PROTON COLLIDER HERA * Albert De Roeck Deutsches Eiektronen-Synchroiron DESY. Hamburg 1 Introduction On the 31st of May 1992 the first electron-proton (ep) collisions were observed in the HI and ZEUS experiments at the newly commissioned high energy collider HERA, in Hamburg. Ger many. HERA is the first electron-proton coEider in the world: 26.7 GeV electrons collide on 820 GeV protons, yielding an ep centre of mass system (CMS) energy of 296 GeV. Already the results from the first data collected by the experiments have given important new information on the structure of the proton, on interactions of high energetic photons with matter and on searches for exotic particles. These lectures give a summary of the physics results obtained by the Hi and ZEUS experiments using the data collected in 1992 and 1993. Electron-proton, or more general lepton-hadron experiments, have been playing a major role in our understanding of the structure of matter for the last 30 years. At the end of the sixties experiments with electron beams on proton targets performed at the Stanford Lin ear Accelerator revealed that the proton had an internal structure.1 It was suggested that the proton consists of pointlike objects, called partons. 2 These partoas were subsequently identified with quarks which until then were only mathematical objects for the fundamen tal representation of the SU(3) symmetry group, used to explain the observed multiplets in hadron spectroscopy. -

Make an Atom Vocabulary Grade Levels
MAKE AN ATOM Fundamental to physical science is a basic understanding of the atom. Atoms are comprised of protons, neutrons, and electrons. Protons and neutrons are at the center of the atom while electrons live in lobe-shaped clouds outside the nucleus. The number of electrons usually matches the number of protons, yielding a net neutral charge for the atom. Sometimes an atom has less neutrons or more neutrons than protons. This is called an isotope. If an atom has different numbers of electrons than protons, then it is an ion. If an atom has different numbers of protons, it is a different element all together. Scientists at Idaho National Laboratory study, create, and use radioactive isotopes like Uranium 234. The 234 means this isotope has an atomic mass of 234 Atomic Mass Units (AMU). GRADE LEVELS: 3-8 VOCABULARY Atom – The basic unit of a chemical element. Proton – A stable subatomic particle occurring in all atomic nuclei, with a positive electric charge equal in magnitude to that of an electron, but of opposite sign. Neutron – A subatomic particle of about the same mass as a proton but without an electric charge, present in all atomic nuclei except those of ordinary hydrogen. Electron – A stable subatomic particle with a charge of negative electricity, found in all atoms and acting as the primary carrier of electricity in solids. Orbital – Each of the actual or potential patterns of electron density that may be formed on an atom or molecule by one or more electrons. Ion – An atom or molecule with a net electric charge due to the loss or gain of one or more electrons.