Haploinsufficiency of TAB2 Causes Congenital Heart
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Tak1 Polyclonal Antibody Catalog # AP72719
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Tak1 Polyclonal Antibody Catalog # AP72719 Specification Tak1 Polyclonal Antibody - Product Information Application IF Primary Accession O43318 Reactivity Human, Mouse, Rat Host Rabbit Clonality Polyclonal Tak1 Polyclonal Antibody - Additional Information Gene ID 6885 Other Names MAP3K7; TAK1; Mitogen-activated protein kinase kinase kinase 7; Transforming growth factor-beta-activated kinase 1; TGF-beta-activated kinase 1 Dilution IF~~IF: 1:50-200 Western Blot: 1/500 - 1/2000. Immunohistochemistry: 1/100 - 1/300. ELISA: 1/40000. Not yet tested in other applications. Format Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide. Storage Conditions -20℃ Tak1 Polyclonal Antibody - Protein Information Name MAP3K7 {ECO:0000303|PubMed:28397838, ECO:0000312|HGNC:HGNC:6859} Function Serine/threonine kinase which acts as an essential component of the MAP kinase signal transduction pathway. Plays an important role in the cascades of cellular responses evoked by changes in the environment. Mediates signal transduction of TRAF6, various cytokines including Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 interleukin-1 (IL-1), transforming growth factor-beta (TGFB), TGFB-related factors like BMP2 and BMP4, toll-like receptors (TLR), tumor necrosis factor receptor CD40 and B-cell receptor (BCR). Ceramides are also able to activate MAP3K7/TAK1. Once activated, acts as an upstream activator of the MKK/JNK signal transduction cascade and the p38 MAPK signal transduction cascade through the phosphorylation and activation of several MAP kinase kinases like MAP2K1/MEK1, MAP2K3/MKK3, MAP2K6/MKK6 and MAP2K7/MKK7. -

Simultaneous Genome-Wide Association Studies of Anti-Cyclic Citrullinated Peptide in Rheumatoid Arthritis Using Penalized Orthogonal-Components Regression
BMC Proceedings BioMed Central Proceedings Open Access Simultaneous genome-wide association studies of anti-cyclic citrullinated peptide in rheumatoid arthritis using penalized orthogonal-components regression Yanzhu Lin1,MinZhang1,LiboWang1, Vitara Pungpapong1, James C Fleet2 and Dabao Zhang*1 Addresses: 1Department of Statistics, Purdue University, West Lafayette, Indiana 47907, USA and 2Department of Foods and Nutrition, Purdue University, West Lafayette, Indiana 47907, USA E-mail: Yanzhu Lin - [email protected]; Min Zhang - [email protected]; Libo Wang - [email protected]; Vitara Pungpapong - [email protected]; James C Fleet - [email protected]; Dabao Zhang* - [email protected] *Corresponding author from Genetic Analysis Workshop 16 St Louis, MO, USA 17-20 September 2009 Published: 15 December 2009 BMC Proceedings 2009, 3(Suppl 7):S20 doi: 10.1186/1753-6561-3-S7-S20 This article is available from: http://www.biomedcentral.com/1753-6561/3/S7/S20 © 2009 Lin et al; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Genome-wide associations between single-nucleotide polymorphisms and clinical traits were simultaneously conducted using penalized orthogonal-components regression. This method was developed to identify the genetic variants controlling phenotypes from a massive number of candidate variants. By investigating the association between all single-nucleotide polymorphisms to the phenotype of antibodies against cyclic citrullinated peptide using the rheumatoid arthritis data provided by Genetic Analysis Workshop 16, we identified genetic regions which may contribute to the pathogenesis of rheumatoid arthritis. -

Wo 2010/075007 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 1 July 2010 (01.07.2010) WO 2010/075007 A2 (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12Q 1/68 (2006.01) G06F 19/00 (2006.01) kind of national protection available): AE, AG, AL, AM, C12N 15/12 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) International Application Number: DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US2009/067757 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 11 December 2009 ( 11.12.2009) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (25) Filing Language: English SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, (26) Publication Language: English TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 12/3 16,877 16 December 2008 (16.12.2008) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (71) Applicant (for all designated States except US): DODDS, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, W., Jean [US/US]; 938 Stanford Street, Santa Monica, TM), European (AT, BE, BG, CH, CY, CZ, DE, DK, EE, CA 90403 (US). -

Genome-Wide Analysis of Host-Chromosome Binding Sites For
Lu et al. Virology Journal 2010, 7:262 http://www.virologyj.com/content/7/1/262 RESEARCH Open Access Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1) Fang Lu1, Priyankara Wikramasinghe1, Julie Norseen1,2, Kevin Tsai1, Pu Wang1, Louise Showe1, Ramana V Davuluri1, Paul M Lieberman1* Abstract The Epstein-Barr Virus (EBV) Nuclear Antigen 1 (EBNA1) protein is required for the establishment of EBV latent infection in proliferating B-lymphocytes. EBNA1 is a multifunctional DNA-binding protein that stimulates DNA replication at the viral origin of plasmid replication (OriP), regulates transcription of viral and cellular genes, and tethers the viral episome to the cellular chromosome. EBNA1 also provides a survival function to B-lymphocytes, potentially through its ability to alter cellular gene expression. To better understand these various functions of EBNA1, we performed a genome-wide analysis of the viral and cellular DNA sites associated with EBNA1 protein in a latently infected Burkitt lymphoma B-cell line. Chromatin-immunoprecipitation (ChIP) combined with massively parallel deep-sequencing (ChIP-Seq) was used to identify cellular sites bound by EBNA1. Sites identified by ChIP- Seq were validated by conventional real-time PCR, and ChIP-Seq provided quantitative, high-resolution detection of the known EBNA1 binding sites on the EBV genome at OriP and Qp. We identified at least one cluster of unusually high-affinity EBNA1 binding sites on chromosome 11, between the divergent FAM55 D and FAM55B genes. A con- sensus for all cellular EBNA1 binding sites is distinct from those derived from the known viral binding sites, sug- gesting that some of these sites are indirectly bound by EBNA1. -

Protein Phosphorylation Sites Notes References Transcription Factors
Protein Phosphorylation Notes References sites Transcription AML1 Ser249, Ser266 Phosphorylation of the acute myelogenous leukemia 1 (AML1) potentially regulates its (Tanaka et al., 1996) Factors (RUNX1) transactivation potential and thereby modifies myeloid hematopoietic differentiation. Androgen Ser514 Phosphorylation of androgen receptor by ERKs is controversial. May play a role in survival (Yeh et al., 1999) receptor and proliferation of prostate cells. Arix/Phox2a Ser36, Ser71 Phosphorylation of Arix by ERK1/2 inhibits its ability to interact with target genes. (Hsieh et al., 2005) ATF2 Thr71 This phosphorylation together with the phosphorylation of Thr69 by p38MAPK induces (Ouwens et al., 2002) activation of ATF2. Beta2 (Neuro Ser274 Phosphorylation of this HLH transcription factor induces its glucose-sensitive transactivation (Khoo et al. 2003) D1) and enhances its capability to heterodimerize with E47/12 and bind to DNA. BCL-6 Ser333, Ser343 Phosphorylation of BCL-6 induces its degradation that is important for B-cell differentiation (Niu et al., 1998) and antibody response. BMAL1 Ser527, Thr534, Phosphorylation of this helix-loop-helix-PAS transcription factor inhibits its activity as a clock (Sanada et al., 2002) Ser599 (chicken) oscillator. CBP Thr188 Phosphorylation of the C-terminal region of the CREB binding protein (CBP, and also its (Janknecht and homolog p300) enhances its transactivation potential. Nordheim, 1996) C/EBPb Thr188 (rat) Phosphorylation of the CCAAT/enhancer-binding protein-b (C/EBPb) by ERK2 (not ERK1) (Hanlon et al., 2001) enhances its interaction with SRF and its transactivation activity. CRY1/2 Ser265, Ser557 Phosphorylation of the Cryptochromes proteins CRY1 and 2 results in attenuation of their (Sanada et al., 2004) (CRY1, mouse) ability to inhibit BMAL1:CLOCK-mediated transcription. -

Functional Genomics Atlas of Synovial Fibroblasts Defining Rheumatoid Arthritis
medRxiv preprint doi: https://doi.org/10.1101/2020.12.16.20248230; this version posted December 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Functional genomics atlas of synovial fibroblasts defining rheumatoid arthritis heritability Xiangyu Ge1*, Mojca Frank-Bertoncelj2*, Kerstin Klein2, Amanda Mcgovern1, Tadeja Kuret2,3, Miranda Houtman2, Blaž Burja2,3, Raphael Micheroli2, Miriam Marks4, Andrew Filer5,6, Christopher D. Buckley5,6,7, Gisela Orozco1, Oliver Distler2, Andrew P Morris1, Paul Martin1, Stephen Eyre1* & Caroline Ospelt2*,# 1Versus Arthritis Centre for Genetics and Genomics, School of Biological Sciences, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, UK 2Department of Rheumatology, Center of Experimental Rheumatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland 3Department of Rheumatology, University Medical Centre, Ljubljana, Slovenia 4Schulthess Klinik, Zurich, Switzerland 5Institute of Inflammation and Ageing, University of Birmingham, Birmingham, UK 6NIHR Birmingham Biomedical Research Centre, University Hospitals Birmingham NHS Foundation Trust, University of Birmingham, Birmingham, UK 7Kennedy Institute of Rheumatology, University of Oxford Roosevelt Drive Headington Oxford UK *These authors contributed equally #corresponding author: [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. 1 medRxiv preprint doi: https://doi.org/10.1101/2020.12.16.20248230; this version posted December 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. -

2219.Full.Pdf
The JNK Are Important for Development and Survival of Macrophages S. Roy Himes, David P. Sester, Timothy Ravasi, Stephen L. Cronau, Tedjo Sasmono and David A. Hume This information is current as of September 28, 2021. J Immunol 2006; 176:2219-2228; ; doi: 10.4049/jimmunol.176.4.2219 http://www.jimmunol.org/content/176/4/2219 Downloaded from References This article cites 64 articles, 40 of which you can access for free at: http://www.jimmunol.org/content/176/4/2219.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 28, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology The JNK Are Important for Development and Survival of Macrophages S. Roy Himes, David P. Sester, Timothy Ravasi, Stephen L. Cronau, Tedjo Sasmono, and David A. Hume1 We report in this study that activation of the JNK by the growth factor, CSF-1 is critical for macrophage development, prolif- eration, and survival. -
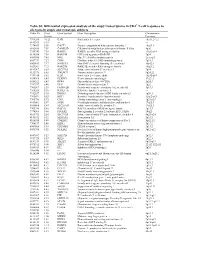
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

MAP3K7 (Phospho-Thr187) Antibody Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP52512
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 MAP3K7 (Phospho-Thr187) Antibody Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP52512 Specification MAP3K7 (Phospho-Thr187) Antibody - Product Information Application WB Primary Accession O43318 Reactivity Human, Mouse, Rat Host Rabbit Clonality Polyclonal Calculated MW 67196 MAP3K7 (Phospho-Thr187) Antibody - Additional Information Gene ID 6885 Other Names Mitogen-activated protein kinase kinase Western blot analysis of extracts from 3T3 kinase 7, Transforming growth factor-beta-activated kinase 1, cells, treated with heat shock, using MAP3K7 TGF-beta-activated kinase 1, MAP3K7, TAK1 (Phospho-Thr187) antibody. Dilution WB~~1:1000 Format Rabbit IgG in phosphate buffered saline (without Mg2+ and Ca2+), pH 7.4, 150mM NaCl, 0.09% (W/V) sodium azide and 50% glycerol. Storage Conditions -20℃ MAP3K7 (Phospho-Thr187) Antibody - Protein Information Western blot analysis of extracts from 3T3 cells (Lane 2), A549 cells (Lane 3) and Hela cells (Lane 4), using MAP3K7 Name MAP3K7 (Phospho-Thr187) Antibody. The lane on the {ECO:0000303|PubMed:28397838, ECO:0000312|HGNC:HGNC:6859} left is treated with synthesized peptide. Function Serine/threonine kinase which acts as an MAP3K7 (Phospho-Thr187) Antibody - essential component of the MAP kinase Background signal transduction pathway. Plays an important role in the cascades of cellular Serine/threonine kinase which acts as an Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 responses evoked by changes in the essential component of the MAP kinase signal environment. Mediates signal transduction transduction pathway. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

TAB2 Antibody Catalog # ASC10267
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 TAB2 Antibody Catalog # ASC10267 Specification TAB2 Antibody - Product Information Application IHC Primary Accession Q9NYJ8 Other Accession NP_055908, 14149669 Reactivity Human Host Rabbit Clonality Polyclonal Isotype IgG Application Notes TAB2 antibody can be used for detection of TAB2 by immunohistoc Immunohistochemistry of TAB2 in human hemistry at 5 brain tissue with TAB2 antibody at 5 µg/mL. µg/mL. TAB2 Antibody - Background TAB2 Antibody - Additional Information TAB2 Antibody: TAB2 is an activator of Gene ID 23118 MAP3K7/TAK1, which is required for for the IL-1 Other Names induced activation NF-kappaB and MAPK8/JNK. TAB2 Antibody: CHTD2, MAP3K7IP2, This protein forms a kinase complex with KIAA0733, TGF-beta-activated kinase 1 and TRAF6, MAP3K7 and TAB1, thus serves as an MAP3K7-binding protein 2, adaptor linking MAP3K7 and TRAF6. This Mitogen-activated protein kinase kinase protein, TAB1, and MAP3K7 also participate in kinase 7-interacting protein 2, TAB-2, the signal transduction induced by TGF-beta activated kinase 1/MAP3K7 TNFSF11/RANKL through the activation of the binding protein 2 receptor activator of NF-kappaB (TNFRSF11A/RANK), which may regulate the Target/Specificity development and function of osteoclasts. TAB2; TAB2 antibody is human specific. Recent experiments have shown that TAB2 and TAB2 antibody is predicted not to cross-react with other TAB proteins. the related protein TAB3 constitutitvely interact with the autophagy mediator Beclin-1; Reconstitution & Storage upon induction of autophagy, these proteins TAB2 antibody can be stored at 4℃ for dissociate from Beclin-1 and bind TAK1. -

The KMT1A-GATA3-STAT3 Circuit Is a Novel Self-Renewal Signaling of Human Bladder Cancer Stem Cells Zhao Yang1, Luyun He2,3, Kais
The KMT1A-GATA3-STAT3 circuit is a novel self-renewal signaling of human bladder cancer stem cells Zhao Yang1, Luyun He2,3, Kaisu Lin4, Yun Zhang1, Aihua Deng1, Yong Liang1, Chong Li2, 5, & Tingyi Wen1, 6, 1CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China 2Core Facility for Protein Research, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China 3CAS Key Laboratory of Infection and Immunity, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China 4Department of Oncology, the Second Affiliated Hospital of Soochow University, Suzhou 215000, China 5Beijing Jianlan Institute of Medicine, Beijing 100190, China 6Savaid Medical School, University of Chinese Academy of Sciences, Beijing 100049, China Correspondence author: Tingyi Wen, e-mail: [email protected] Chong Li, e-mail: [email protected] Supplementary Figure S1. Isolation of human bladder cancer stem cells. BCMab1 and CD44 were used to isolate bladder cancer stem cells (BCSCs: BCMab1+CD44+) and bladder cancer non-stem cells (BCNSCs: BCMab1-CD44-) from EJ, samples #1 and #2 by flow cytometry. Supplementary Figure S2. Gene ontology analysis of downregulated genes of human BCSCs. (A) Pathway enrichment of 103 downregulated genes in BCSCs. (B) The seven downregulated genes in BCSCs participating in centromeric heterochromatin, mRNA-3’-UTR binding and translation regulator activity signaling pathways were validated by qRT-PCR. Data are presented as mean ± SD. P < 0.05; P < 0.01. Supplementary Figure S3. The expression of KMT1A is higher in human BC than that in peri-tumor tissues. (A) The expression of KMT1A was higher in BC samples than that in peri-tumors as assessed by immunohistochemistry, Scale bar = 50 m.