Juan Antonio Rodríguez Pérez
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
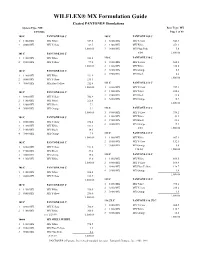
WILFLEX® MX Formulation Guide
WILFLEX® MX Formulation Guide Coated PANTONE® Simulations System Type: MX Base Type: MX 3/29/2006 Page 1 of 83 100 C PANTONE 100 C 109 C PANTONE 109 C 1 11888MX MX White 937.5 1 88888MX MX Yellow 543.9 2 88888MX MX Yellow 62.5 2 11888MX MX White 451.1 1,000.00 3 98880MX MX Fluo Pink 5.0 101 C PANTONE 101 C 8/04 1,000.00 1 11888MX MX White 902.8 110 C PANTONE 110 C 2 88888MX MX Yellow 97.2 1 88888MX MX Yellow 660.1 1,000.00 2 11888MX MX White 330.0 102 C PANTONE 102 C 3 38888MX MX Orange 5.5 4 19888MX MX Black 4.4 1 11888MX MX White 531.9 1,000.00 2 88888MX MX Yellow 255.3 3 98888MX MX Fluo Yellow 212.8 111 C PANTONE 111 C 1,000.00 1 88888MX MX Yellow 727.1 103 C PANTONE 103 C 2 11888MX MX White 238.4 3 19888MX MX Black 23.8 1 88888MX MX Yellow 762.9 4 38888MX MX Orange 10.7 2 11888MX MX White 226.8 1,000.00 3 19888MX MX Black 7.2 4 38888MX MX Orange 3.1 112 C PANTONE 112 C 1,000.00 1 88888MX MX Yellow 936.2 104 C PANTONE 104 C 2 11888MX MX White 21.3 3 19888MX MX Black 23.4 1 88888MX MX Yellow 874.4 4 38888MX MX Orange 19.1 2 11888MX MX White 103.3 8/04 1,000.00 3 19888MX MX Black 14.3 4 38888MX MX Orange 7.9 113 C PANTONE 113 C 1,000.00 1 11888MX MX White 867.1 105 C PANTONE 105 C 2 88888MX MX Yellow 132.1 3 38888MX MX Orange 0.8 1 88888MX MX Yellow 921.6 1/14/04 1,000.00 2 19888MX MX Black 39.2 3 38888MX MX Orange 19.6 114 C PANTONE 114 C 4 11888MX MX White 19.6 1 11888MX MX White 693.5 1,000.00 2 88888MX MX Yellow 166.4 106 C PANTONE 106 C 3 98888MX MX Fluo Yellow 138.7 4 38888MX MX Orange 1.4 1 11888MX MX White 804.3 1,000.00 2 88888MX -

Simulations of PANTONE MATCHING SYSTEM Colors
Simulations of PANTONE MATCHING SYSTEM colors PANTONE Process PANTONE Purple C PANTONE Process PANTONE PANTONE 100 C PANTONE 106 C PANTONE 113 C PANTONE 120 C PANTONE 1205 C PANTONE 127 C PANTONE 134 C PANTONE 1345 C PANTONE 141 C PANTONE 148 C PANTONE 1485 C PANTONE 155 C PANTONE 1555 C PANTONE 162 C PANTONE 1625 C PANTONE 169 C PANTONE 176 C PANTONE 1765 C PANTONE 1767 C PANTONE 182 C PANTONE 189 C PANTONE 1895 C PANTONE 196 C PANTONE 203 C PANTONE 210 C PANTONE 217 C PANTONE 223 C PANTONE 230 C PANTONE 236 C Yellow C Yellow C HEXACHROME Yellow C PANTONE Yellow 012 C PANTONE Violet C PANTONE Process PANTONE PANTONE 101 C PANTONE 107 C PANTONE 114 C PANTONE 121 C PANTONE 1215 C PANTONE 128 C PANTONE 135 C PANTONE 1355 C PANTONE 142 C PANTONE 149 C PANTONE 1495 C PANTONE 156 C PANTONE 1565 C PANTONE 163 C PANTONE 1635 C PANTONE 170 C PANTONE 177 C PANTONE 1775 C PANTONE 1777 C PANTONE 183 C PANTONE 190 C PANTONE 1905 C PANTONE 197 C PANTONE 204 C PANTONE 211 C PANTONE 218 C PANTONE 224 C PANTONE 231 C PANTONE 237 C Magenta C HEXACHROME Orange C PANTONE Orange 021 PANTONE Blue 072 C PANTONE Process PANTONE PANTONE 102 C PANTONE 108 C PANTONE 115 C PANTONE 122 C PANTONE 1225 C PANTONE 129 C PANTONE 136 C PANTONE 1365 C PANTONE 143 C PANTONE 150 C PANTONE 1505 C PANTONE 157 C PANTONE 1575 C PANTONE 164 C PANTONE 1645 C PANTONE 171 C PANTONE 178 C PANTONE 1785 C PANTONE 1787 C PANTONE 184 C PANTONE 191 C PANTONE 1915 C PANTONE 198 C PANTONE 205 C PANTONE 212 C PANTONE 219 C PANTONE 225 C PANTONE 232 C PANTONE 238 C C Cyan C HEXACHROME Magenta -

British American Tobacco V Department of Health Judgment
Neutral Citation Number: [2016] EWHC 1169 (Admin) Case No: CO/2322/2015, CO/2323/2015, CO/2352/2015, CO/2601/2015 & CO/2706/2015 IN THE HIGH COURT OF JUSTICE QUEEN'S BENCH DIVISION ADMINISTRATIVE COURT Royal Courts of Justice Strand, London, WC2A 2LL Date: 19/05/2016 Before : MR JUSTICE GREEN - - - - - - - - - - - - - - - - - - - - - Between : THE QUEEN On the application of (1) BRITISH AMERICAN TOBACCO (UK) LIMITED (2) BRITISH AMERICAN TOBACCO (BRANDS) INC. (3) BRITISH AMERICAN TOBACCO (INVESTMENTS) LIMITED First Claimants - and - SECRETARY OF STATE FOR HEALTH Defendant And Between : THE QUEEN On the application of (1) PHILIP MORRIS BRANDS SARL (2) PHILIP MORRIS PRODUCTS SA Second (3) PHILIP MORRIS LIMITED Claimants - and - SECRETARY OF STATE FOR HEALTH Defendant And Between : THE QUEEN On the application of (1) JT INTERNATIONAL SA Third (2) GALLAHER LIMITED Claimants Judgment Approved by the court for handing down. Tobacco Packaging - and - SECRETARY OF STATE FOR HEALTH Defendant And Between : THE QUEEN On the application of Fourth IMPERIAL TOBACCO LIMITED Claimant - and - SECRETARY OF STATE FOR HEALTH Defendant ACTION ON SMOKING AND HEALTH (“ASH”) Intervener - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - Case No: CO/2322/2015: NIGEL PLEMING QC, GEOFFREY HOBBS QC, DAVID SCANNELL and PHILIP ROBERTS (instructed by Herbert Smith Freehills LLP) appeared on behalf of the First Claimants Case No: CO/2323/2015: MARIE DEMETRIOU QC and DANIEL PICCININ (instructed by Skadden, Arps, Slate, Meagher & Flom (UK) -

Paper Template
SESUG 2016 Paper RV-180 Color Speaks Louder than Words Jaime Thompson, Zencos Consulting ABSTRACT What if color blind people saw real colors and it was everyone else who's color blind? Imagine finishing a Rubik's cube wondering why people find it so difficult. Or relying on the position, rather than the color of a traffic light in determining when to stop and go. Color matters! Color enhances our perspective, it can change how we feel, but it also varies culturally. In western countries, red and white have opposing symbolic meanings to eastern countries which in turn can send the wrong message if misused. Color can fundamentally change a report, therefore, finding the right color palettes for data visualizations is essential. In this paper, I will cover the significance of color and how to pick a palette for your next SAS® Visual Analytics report. INTRODUCTION Color is used in the workplace to improve performance and productivity. Color is used in marketing to establish identity. Color is used in paintings to communicate emotion. Color is used in stores to sell products. Color is everywhere and generally applied purposely. The government of Australia spent several months researching to find the worlds ugliest color. Why? They were looking for a color so distasteful that if it was used for packaging tobacco products it would deter people from smoking [7]. Just as the Australian government needed to pick the right color for a specific job, it is essential to select the right colors for your data visualization so their meanings are conveyed and interpreted correctly. -

Plain Packaging Update
Plain Packaging Update March 2020 www.galalaw.com Argentina about the active ingredient, whether the product is by present, there are no legislative plans to introduce such requirements. Union” for additional requirements.) Since 2017, health India products. For example, the advertising of tobacco products is Peru South Africa cover 50% of both the front and back sides of the packaging), Venezuela To date, the discussion regarding plain packaging has been prescription, etc., shall be provided on the package). However, See “European Union” for additional requirements. messages must be added to advertising for manufactured food Plain packaging is not required in India. However, India has prohibited on television, radio, cinemas, and other broadcasting Although Peru does not have any plain packaging requirements, In May 2018, the South African government’s Department of alcohol, medicines, and some others. On 28 January 2020, Although plain packaging is not currently required in Venezuela, limited to tobacco products. So far, Argentina has not enacted there are no prohibitions on placing branding or logos on and drinks products on television, cinema, radio, newspapers, taken major anti-tobacco initiatives in recent years, such as the media, and health warnings and graphics must cover 65% of under Peruvian food law, it is not possible to use children's Health released the Draft Control of Tobacco Products and an anti-tobacco bill was submitted to the Ukrainian Parliament. graphic warnings must be 100% over one side, and 30% on the a plain packaging law. While several proposals for tobacco product packaging. Czech Republic and on posters and marketing flyers distributed in supermarkets. -

Sunmatch Solvent-Based Inks
SunMatch™ Solvent-Based Inks C37, C70, HG. PO, YN, Z, ZGL Series Formula Guide to Simulate Pantone® Colors Sun Chemical The PANTONE MATCHING SYSTEM® Trademark and Copyright Notice Sun Chemical’s color-mixing ink formulations for screen-process printing produce only simulations of PANTONE Colors in this color reproduction method due to differences in ink film, opacity and pigment selection. The pigment selection used in blending screen-process inks may cause metamerism. Pantone, Inc. assumes no responsibility for formula accuracy. All trademarks used herein are either the property of Sun Chemical, Pantone, Inc. or other companies. PANTONE® is a trademark of PANTONE, Inc. Portions © PANTONE, Inc., 1991. A GUIDE TO SIMULATION OF PANTONE COLORS WITH SCREEN INKS This formulating guide is designed to assist the screen printer in simulating PANTONE Colors as accurately as possible with screen inks. However, there are many factors such as mesh, substrate color and texture, which may affect the color achieved with a screen printing ink. It is therefore recommended that a small quantity of ink is first mixed, checked and adjusted, if necessary, prior to mixing bulk quantities. IMPORTANT The commitment of Sun Chemical to quality assurance ensures that every effort is made to supply inks which match the colors illustrated. However as differences in substrates and application conditions can affect the color achieved, all inks are supplied without warranty, either expressed or implied. The relevant Product Data Sheet should be consulted before using any range of inks. Due to differences in opacity, ink film and colorants used, the colors shown are simulations of the respective PANTONE-identified solid color standards. -
全文本) Acceptance for Registration (Full Version)
公報編號 Journal No.: 645 公布日期 Publication Date: 14-08-2015 分項名稱 Section Name: 接納註冊 (全文本) Acceptance for Registration (Full Version) 香港特別行政區政府知識產權署商標註冊處 Trade Marks Registry, Intellectual Property Department The Government of the Hong Kong Special Administrative Region 接納註冊 (全文本) 商標註冊處處長已根據《商標條例》(第 559 章)第 42 條,接納下列商標的註冊申請。現根據《商標條 例》第 43 條及《商標規則》(第 559 章附屬法例)第 15 條,公布申請的詳情。 根據《商標條例》第 44 條及《商標規則》第 16 條,任何人擬就下列商標的註冊提出反對,須在本公告 公布日期起計的三個月內,採用表格第 T6 號提交反對通知。(例如,若果公布日期爲 2003 年 4 月 4 日,則該三個月的最後一日爲 2003 年 7 月 3 日。)反對通知須載有反對理由的陳述及《商標規則》第 16(2)條所提述的事宜。反對人須在提交反對通知的同時,將該通知的副本送交有關申請人。 有關商標註冊處處長根據商標條例(第 43 章)第 13 條/商標條例(第 559 章)附表 5 第 10 條所接納的註冊申 請,請到 http://www.gld.gov.hk/cgi-bin/gld/egazette/index.cgi?lang=c&agree=0 檢視電子憲報。 ACCEPTANCE FOR REGISTRATION (FULL VERSION) The Registrar of Trade Marks has accepted the following trade marks for registration under section 42 of the Trade Marks Ordinance (Cap. 559). Under section 43 of the Trade Marks Ordinance and rule 15 of the Trade Marks Rules (Cap. 559 sub. leg.), the particulars of the applications are published. Under section 44 of the Trade Marks Ordinance and rule 16 of the Trade Marks Rules, any person who wishes to oppose the registration of any of these marks shall, within the 3-month period beginning on the date of this publication, file a notice of opposition on Form T6. (For example, if the publication date is 4 April 2003, the last day of the 3-month period is 3 July 2003.) The notice of opposition shall include a statement of the grounds of opposition and the matters referred to in rule 16(2). -

Vennligblad 22.6
Vennligblad Friendly pages for the ‘friendly people’ of Vennligfolk and their friends 2018 Officers Sons of Norway Lodge #5-627 for Central Wisconsin, President Susan Morton Stevens Point, Whiting and Plover, Wisconsin (715)341-7248 [email protected] VOLUME 25 ISSUE 6 NOVEMBER OG DESEMBER 2018 Vice President Lois Hagen (715)344-7460 [email protected] Treasurer Judy Pesanka (715)344-0719 msbrewer2charter.net ne of the greatest pleasures of belonging of these elections and the Financial Secretary Arno Morton O to Vennligfolk Lodge is the people we people running for office. (715)341-7248 have met who have become dear friends. One November is also the time [email protected] of these dear friends is charter member Ruth for the election of Secretary Harpstead. Ruth, along with her husband, Vennligfolk officers. You Bea Berg Milo, have been the “heart” of Vennligfolk won’t be seeing or hearing (715)544-4490 [email protected] for as long as I have been a member. While campaign ads for the peo- Program Director Milo was lodge president for fourteen years, ple on the ballot for our Joyce Polson Ruth was the “First Lady” of the lodge. Re- election, but you will see a list of their names (715)341-4545 cently, while I was going through some pic- in this issue of Vennligblad. I am so appre- [email protected] tures of Ruth that I have on my phone, I came ciative of the wonderful people I have had Newsletter Editor across the group picture we had taken at last the pleasure of working with this last year Marv Lang (715)341-3201 year’s Julefest. -

Sunmatch Pantone Formulas Guide -U.V.Curable Inks Com FLX GEN7
SunMatch™ UV Curable Inks for Display and Hi-Tech Automotive Graphic Applications COMETAL, FLX, GEN7, GLS, MRV, PD, REV, UMP, UVN, VAC, VYB Series Formula Guide to Simulate Pantone® Colors Sun Chemical The PANTONE MATCHING SYSTEM® Trademark and Copyright Notice Sun Chemical’s color-mixing ink formulations for screen-process printing produce only simulations of PANTONE Colors in this color reproduction method due to differences in ink film, opacity and pigment selection. The pigment selection used in blending screen-process inks may cause metamerism. Pantone, Inc. assumes no responsibility for formula accuracy. All trademarks used herein are either the property of Sun Chemical, Pantone, Inc. or other companies. PANTONE® is a trademark of PANTONE, Inc. Portions © PANTONE, Inc., 1991. A GUIDE TO SIMULATION OF PANTONE COLORS WITH SCREEN INKS This formulating guide is designed to assist the screen printer in simulating PANTONE Colors as accurately as possible with screen inks. However, there are many factors such as mesh, substrate color and texture, which may affect the color achieved with a screen printing ink. It is therefore recommended that a small quantity of ink is first mixed, checked and adjusted, if necessary, prior to mixing bulk quantities. IMPORTANT The commitment of Sun Chemical to quality assurance ensures that every effort is made to supply inks which match the colors illustrated. However as differences in substrates and application conditions can affect the color achieved, all inks are supplied without warranty, either expressed or implied. The relevant Product Data Sheet should be consulted before using any range of inks. Due to differences in opacity, ink film and colorants used, the colors shown are simulations of the respective PANTONE-identified solid color standards. -

MADEIRA Thread Colour Guide
MADEIRA Thread Colour Guide The below pantone colour swatches are indicative only and a best match for the thread colour from the Polyneon range. PMS colours viewed on screen and printed on paper are approximate only. To search for a requested colour, please activate the Acrobat search function by pressing “Ctrl F”. PANTONE MADEIRA PANTONE MADEIRA Matching System Colour Matching System Colour White 1801 PANTONE 116 C 1824 Black 1800 PANTONE 117 C 1792 PANTONE Cool Gray 1 C 1686 PANTONE 119 C 1793 PANTONE Cool Gray 4 C 1687 PANTONE 121 C 1735 PANTONE Cool Gray 5 C 1812 PANTONE 122 C 1683 PANTONE Cool Gray 6 C 1505 PANTONE 123 C 1624 PANTONE Cool Gray 7 C 1613 PANTONE 124 C 1772 PANTONE Cool Gray 8 C 1918 PANTONE 125 C 1672 PANTONE Cool Gray 9 C 1689 PANTONE 127 C 1866 PANTONE Cool Gray 10 C 1575 PANTONE 1205 c 1622 PANTONE Cool Gray 11 C 1540 PANTONE 1235 C 1951 PANTONE Black 5 C 1558 PANTONE 1245 C 1791 PANTONE Black 7 C 1560 PANTONE 130 C 1951 PANTONE Process Blue C 1977 PANTONE 131 C 1725 PANTONE Reflex Blue C 1766 PANTONE 136 C 1625 PANTONE Blue 072 C 1767 PANTONE 1365 C 1923 PANTONE Orange 021 C 1678 PANTONE 137 C 1955 PANTONE Warm Red C 1588 PANTONE 1375 C 1755 PANTONE Red 032 C 1734 PANTONE 138 C 1763 PANTONE Yellow 012 C 1924 PANTONE 1395 C 1730 PANTONE 100 C 1735 PANTONE 140 C 1906 PANTONE 102 C 1924 PANTONE 141 C 1771 PANTONE 106 C 1727 PANTONE 142 C 1771 PANTONE 107 C 1924 PANTONE 143 C 1951 PANTONE 108 C 1924 PANTONE 144 C 1763 PANTONE 109C 1735 PANTONE 148 C 1870 PANTONE 112C 1759 PANTONE 1495 C 1965 PANTONE 114 C 1735 PANTONE 150 C 1755 PANTONE 115 C 1980 PANTONE 155 C 1738 MADEIRA Thread Colour Guide The below pantone colour swatches are indicative only and a best match for the thread colour from the Polyneon range. -

MADEIRA Thread Colour Guide
MADEIRA Thread Colour Guide The below pantone colour swatches are indicative only and a best match for the thread colour from the Polyneon range. PMS colours viewed on screen and printed on paper are approximate only. To search for a requested colour, please activate the Acrobat search function by pressing “Ctrl F”. PANTONE MADEIRA PANTONE MADEIRA Matching System Colour Matching System Colour White 1801 PANTONE 116 C 1824 Black 1800 PANTONE 117 C 1792 PANTONE Cool Gray 1 C 1686 PANTONE 119 C 1793 PANTONE Cool Gray 4 C 1687 PANTONE 121 C 1735 PANTONE Cool Gray 5 C 1812 PANTONE 122 C 1683 PANTONE Cool Gray 6 C 1505 PANTONE 123 C 1624 PANTONE Cool Gray 7 C 1613 PANTONE 124 C 1772 PANTONE Cool Gray 8 C 1918 PANTONE 125 C 1672 PANTONE Cool Gray 9 C 1689 PANTONE 127 C 1866 PANTONE Cool Gray 10 C 1575 PANTONE 1205 c 1622 PANTONE Cool Gray 11 C 1540 PANTONE 1235 C 1951 PANTONE Black 5 C 1558 PANTONE 1245 C 1791 PANTONE Black 7 C 1560 PANTONE 130 C 1951 PANTONE Process Blue C 1977 PANTONE 131 C 1725 PANTONE Reflex Blue C 1766 PANTONE 136 C 1625 PANTONE Blue 072 C 1767 PANTONE 1365 C 1923 PANTONE Orange 021 C 1678 PANTONE 137 C 1955 PANTONE Warm Red C 1588 PANTONE 1375 C 1755 PANTONE Red 032 C 1734 PANTONE 138 C 1763 PANTONE Yellow 012 C 1924 PANTONE 1395 C 1730 PANTONE 100 C 1735 PANTONE 140 C 1906 PANTONE 102 C 1924 PANTONE 141 C 1771 PANTONE 106 C 1727 PANTONE 142 C 1771 PANTONE 107 C 1924 PANTONE 143 C 1951 PANTONE 108 C 1924 PANTONE 144 C 1763 PANTONE 109C 1735 PANTONE 148 C 1870 PANTONE 112C 1759 PANTONE 1495 C 1965 PANTONE 114 C 1735 PANTONE 150 C 1755 PANTONE 115 C 1980 PANTONE 155 C 1738 MADEIRA Thread Colour Guide The below pantone colour swatches are indicative only and a best match for the thread colour from the Polyneon range. -

Raw Umber – It’S Ugly and We Love It
Raw Umber – It’s Ugly and We Love It In 2016, Pantone 448 C was declared as the world’s ugliest color. So ugly, in fact, an Australian research and marketing project covered cigarette packs in the color and successfully discouraged people from smoking. While most people may be disgusted with this green/brown color, often called “Opaque Couché,” we in the scenic art biz call it Raw Umber – and we are madly in love with it! Pantone 448C – The World’s Ugliest Color Rosco’s Off Broadway Raw Umber is one of the few colors most scenic, and prop artists will use straight out the can – no mixing necessary! Once it’s thinned out with water, this paint makes almost anything look old, dirty, and gross with just a pass or two of thin washes and spatter. Because this hue looks like a readily available earth tone, I’m often asked, “isn’t this one of those colors I can pick up at the local paint store?” I have to say that I am unimpressed with the results. Above is a comparison that illustrates the difference between the house paint match and the original Off Broadway Raw Umber. It kind of works, but it ain’t nearly as sexy and I don’t find the house paint as reliable as the Off Broadway. I painted these large vases for the Denver Center in 2007. Note how the drips & washes of Raw Umber add age to the vases and bring out their dimensionality. I also put the Raw Umber to work painting the old, grungy sidewalk set below for Penumbra Theatre Scenic Elements.