THE JOURYAL of Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Carbonylation Reaction of Organoboranes in the Presence of Lithium Triethylborohydride
W&M ScholarWorks Dissertations, Theses, and Masters Projects Theses, Dissertations, & Master Projects 1975 The Carbonylation Reaction of Organoboranes in the Presence of Lithium Triethylborohydride Robert Cloyd Shiffer College of William & Mary - Arts & Sciences Follow this and additional works at: https://scholarworks.wm.edu/etd Part of the Organic Chemistry Commons Recommended Citation Shiffer, Robert Cloyd, "The Carbonylation Reaction of Organoboranes in the Presence of Lithium Triethylborohydride" (1975). Dissertations, Theses, and Masters Projects. Paper 1539624896. https://dx.doi.org/doi:10.21220/s2-z8kr-ve76 This Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Dissertations, Theses, and Masters Projects by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. THE CARBONYLATION REACTION OF ORGANOBORANES U IN THE PRESENCE OF LITHIUM TRIETHYLBOROHYDRIDE A Thesis Presented to The Faculty of the Department of Chemistry The College of William and Mary in Virginia In Partial Fulfillment Of the Requirements for the Degree of Master of Arts by Robert C. Shiffer, Jr. 1975 To my family 61553 9 APPROVAL SHEET This thesis is submitted in partial fulfillment of the requirements for the degree of Master of Arts Robert C. Shaffer Approved, January 1975 Randolph A. Coleman, Ph.D. Robert A. Orwoll, Ph.D. Melvyn D. £> chi ave 1 li, Ph. D. iii TABLE OF CONTENTS ACKNOWLEDGMENT ................................. vi LIST OF TABLES ............ vii LIST OF FIGURES . ............ viii ABSTRACT ................................. ix Chapter I. INTRODUCTION................................... 2 II. THE CARBONYLATION OF B-ALKYL-9-BBN DERIVATIVES IN THE PRESENCE OF' LITHIUM TRIETHYLBOROHYDRIDE OXIDATION OF THE REACTION INTERMEDIATES TO THE CORRESPONDING ALDEHYDES ; .................................. -
Boranes Bases
Boranes Bases Specialty Boron Reagents and Organic Bases Product Overview Enabling the World’s Most Important Products from Lab Through Commercialization At Callery, we enable our customer’s success. From boranes to alkoxides, to custom organometallics, Callery’s impressive combination of innovation, scale, customer service, and supply chain security gives us a distinct advantage. Our products have enabled some of the world’s most successful innovations from billion-dollar blockbuster drugs to next-generation OLEDs. A CLEAR LEADER IN HIGHLY-REACTIVE REAGENTS Our decades of experience and ability to produce highly reactive compounds that begin in our 50,000-ft2 research and development lab to full production of multi tons of product. Combine this with our signature customer support, quality management system, and global logistics expertise, Callery understands what it truly means to de-risk your supply chain. Superior Quality Absolute Security of Supply and Performance and Global Rapid Delivery We continue to deliver high quality reagents, enabling our Callery prides itself in not only providing a quality product, customer’s downstream products and providing distinct but providing a clear pathway to supply. Callery’s North advantages in terms of purity, yield, and overall performance. American location and strict adherence to quality, EHS, Our quality management system means strict adherence to and ISO and EPA standards means we are in control of procedures and a robust change control process. everything from incoming raw materials to shipping of your product. With significant global logistics expertise, including temperature-controlled storage and warehousing facilities in Unparalleled the US and EU, we ensure safe, secure and timely delivery of Customer Support your orders. -

Aluminium Hydrides and Borohydrides
ALUMINIUM HYDRIDES AND BOROHYDRIDES www.acros.com Contents 1 - Introduction . 1 2 - Synthesis and Properties . 2 3 - Chemistry . 5 3.1 Alkylaluminium compounds as Ziegler-Natta co-catalysts . 5 3.2 Complex aluminium hydrides and borohydrides as reducing agents . 7 3.3 The parent compounds lithium aluminium hydride and sodium borohydride . 8 3.3.1. Lithium aluminium hydride. 8 3.3.2. Sodium borohydride and Sodium borodeuteride . 12 3.4 Tuning of the reactivity by different substituents: Derivatives with different sterical and electronical properties . 16 3.5 Modified borohydrides . 17 3.5.1. Lithium borohydride . 17 3.5.2. Potassium borohydride . 18 3.5.3. Tetraalkylammonium- and tetraalkylphosphonium borohydrides . 18 3.5.4. Calcium borohydride . 19 3.5.5. Sodium cyanoborohydride . 19 3.5.6. Sodium triacetoxyborohydride . 22 3.5.7. Tetramethylammonium triacetoxyborohydride (NEW: AO 39285) . 23 3.6 Alkoholate modified aluminium hydrides . 25 3.6.1. Lithium triethoxyaluminium hydride . 25 3.6.2. Lithium tri-tert-butoxyaluminohydride . 25 3.6.3 Sodium bis(2-methoxyethoxy)aluminiumhydride (“SMEAH”) . 25 3.7 Alkylsubstituted borohydrides . 26 3.7.1. Triethylborohydrides . 26 3.7.2. Tri sec-butylborohydrides and trisamylborohydrides . 29 3.7.3. Lithium trisamylborohydride . 30 3.8. Alkylsubstituted aluminiumhydrides . 30 Index . 36 1 - Introduction The importance of organo-boron and organo-aluminium compounds for science and technology has resulted in three Nobel-prizes for Herbert C. Brown for his work on hydroboration1, for Giulio 1Natta -

Ep 1201722 A1
Europäisches Patentamt *EP001201722A1* (19) European Patent Office Office européen des brevets (11) EP 1 201 722 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C09J 4/00, C08F 220/18, 02.05.2002 Bulletin 2002/18 C08F 4/52 (21) Application number: 00650166.2 (22) Date of filing: 23.10.2000 (84) Designated Contracting States: • Coughlan, Gerard AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU Tallaght, Dublin 23, Ireland (GB) MC NL PT SE Designated Extension States: (74) Representative: AL LT LV MK RO SI Parkes, Andrew John Aykroyd et al c/o Tomkins & Co. (71) Applicant: Loctite (R & D) Limited 5 Dartmouth Road Tallaght, Dublin 24 (IE) Dublin 6 (IE) (72) Inventors: • Kneafsey, Brendan Co. Dublin, Ireland (GB) (54) Polymerisation initiators, polymerisable compositions, and uses thereof (57) Metal alkyl borohydrides are used as initiators In particular, alkali metal trialkyl borohydrides are of polymerisation, particularly in adhesive compositions used, the alkali metal salt being selected from the group for bonding a wide range of substrates including low sur- consisting of: Lithium triethylborohydride, Sodium tri- face energy substrates such as polyolefins. As de- ethylborohydride, Potassium triethylborohydride, Lithi- scribed, the metal alkyl borohydrides are of the formula I um tri-sec-butylborohydride, Sodium tri-sec-butylboro- hydride, Potassium tri-sec-butylborohydride, Lithium 9-borabicyclo [3.3.1]-nonane (9BBN) hydride, Lithium thexylborohydride, Lithium trisiamylborohydride, Potas- sium trisiamylborohydride and Lithium triethylborodeu- teride. A polymerisable composition, particularly a two part adhesive composition. comprises: a) a free-radically polymerisable monomer compo- wherein nent, and b) an effective amount of an initiator system for in- 1 R is C1 - C10 alkyl, itiating polymerisation of the free-radically polymer- R2,R3and R4, which may be the same or different, isable monomer, said initiator system comprising a are H, D, C1 -C10 alkyl or C1 -C10 cycloalkyl, phenyl, metal alkyl borohydride. -

Durham E-Theses
Durham E-Theses Studies on the organic chemistry of boron Livingstone, J.G. How to cite: Livingstone, J.G. (1961) Studies on the organic chemistry of boron, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/9111/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk (11 ft u 4 SIAO rsi ^> V 3^NVJ-i3>Vc)V3 Nl 39IMVH3 0 6> CM 5 li 70 7D 0 0> 6 ID 8 79 730 Uoi 0 STUDIES ON THE ORGANIC CHEMISTRY OF BORON BY J. Gr« LIVINGSTONE A dissertation submitted for the Degree of Doctor of Philosophy in the Durham Colleges of the University of Durham. 1961 CONTENTS Page Acknowledgments (1) Memorandum (II) Summary (Ill) FART I Introduction Page Organor-Derivatives of Boric Acid I Trialkyl- and Triaryl Boranes 8 Mixed Trialkyl and Aryl-Alkylboranes 40 Cyclic Boranes 43 Boronic Acids and Esters 46 Borinic Acids and Esters 54 Boroxines 62 Alkyl- and Aryl- Dihaloboranes 65 Dialkyl. -

Recent Developments in Bisdiborane Chemistry: B—C—B, B—C—C—B, B—C=C—B and B—CC—B Compounds
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/264295611 Recent Developments in Bisdiborane Chemistry: B—C—B, B—C—C—B, B—C=C—B and B—CC—B Compounds Article in ChemInform · August 2003 DOI: 10.1002/chin.200332254 CITATIONS READS 0 28 3 authors, including: Valery Dembitsky Hijazi Abu Ali Institute of Marine Biology Birzeit University 341 PUBLICATIONS 4,825 CITATIONS 58 PUBLICATIONS 331 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: Antitumour natural metabolites isolated from fungi, plant and microorganisms View project Plasmalogens of marine invertebrates, functions and structures View project All content following this page was uploaded by Valery Dembitsky on 28 January 2015. The user has requested enhancement of the downloaded file. All in-text references underlined in blue are added to the original document FADA - Birzeit University and are linked to publications on ResearchGate, letting you access and read them immediately. provided by View metadata, citation and similar papers at core.ac.uk CORE brought to you by APPLIED ORGANOMETALLIC CHEMISTRY Appl. Organometal. Chem. 2003; 17: 327–345 Main Group Metal Compounds Published online in Wiley InterScience (www.interscience.wiley.com). DOI:10.1002/aoc.430 Review Recent developments in bisdiborane chemistry: B–C–B, B–C–C–B, B–C C–B and B–C C–B compounds† Valery M. Dembitsky, Hijazi Abu Ali and Morris Srebnik* Department of Medicinal Chemistry & Natural Products, School of Pharmacy, PO Box 12065, Hebrew University of Jerusalem, Jerusalem 91120, Israel Received 18 November 2002; Revised 27 November 2002; Accepted 13 January 2003 An overview of the development of new strategies in organic synthesis with a minimum of chemical steps is becoming increasingly necessary for the efficient assembly of complex molecular structures. -

Chemical Reactivity Evaluation Tool Help Guide
RMT Help Guide v1.0 January 2012 CChheemmiiccaall RReeaaccttiivviittyy EEvvaalluuaattiioonn TTooooll HHeellpp GGuuiiddee Edward M Davis Daniel E Sliva John F. Murphy Robert W. Johnson Steven W. Rudy Copyright © 2012 AIChE P a g e | 1 RMT Help Guide v1.0 January 2012 Table of Contents Follow links ending with an * to a more detailed sectional table of contents. READ ME FIRST ..............................................................................................................3 Acknowledgments .............................................................................................................................5 Legal Disclaimer and Limits of Liability ...............................................................................................7 Preface ...............................................................................................................................................8 Quick Start .......................................................................................................................10 1 Introduction ...................................................................................................................11 1.1 Background ............................................................................................................................... 12 1.2 Objectives ................................................................................................................................. 13 1.3 Limitations ............................................................................................................................... -

Reactions of Organoboranes
AN ABSTRACT OF THE THESIS OF Thomas Myron Thompson for the M. S. in Chemistry (Organic) (Name) (Degree) (Major) Date thesis is presented October 14, 1966, Title REACTIONS OF ORGANOBORANES Abstract approved Redacted for Privacy (Major professor) An examination was made of some reactions involving a trans- fer of carbon from boron to carbon. In the case of the reaction of a C alkylorganoborane with the intermediate was n dichlorocarbene a chloroalkylorganoborane. Oxidation of Cn +1 this intermediate yielded an homologous acid (33 %) and the usual alcohol (66 %). The results of several other transfer studies are presented, and the role of ethereal solvents discussed. REACTIONS OF ORGANOBORANES by THOMAS MYRON THOMPSON A THESIS submitted to OREGON STATE UNIVERSITY in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE June 1967 APPROVED: Redacted for Privacy Assistant Professor of Chemistry In Charge of Major Redacted for Privacy Chairman of Department of Chemistry Redacted for Privacy Dean of Graduate School Date thesis is presented October 14, 1966 Typed by Opal Grossnicklaus ACKNOWLEDGEMENT The author wishes to express his most sincere appreciation to Dr. F. T. Bond. Without his continued inspiration and involvement this thesis could not have been possible. TABLE OF CONTENTS INTRODUCTION 1 DISCUSSION 17 ®CX3 Reactions of Organoboranes with 17 Protonolysis Studies 24 e O Reactions of Organoboranes with CH2- S- (CH3)2 27 Reactions of Organoboranes with Simmons -Smith Reagent 28 EXPERIMENTAL 31 Trihexylborane4 31 T rioctylborazze4 32 Bis(3- methyl -2- butyl) borane 6 32 Bis(3- methyl -2- butyl)octylborane 33 Bis(exo -bicyclo[ 2. -

Tri-N-Butylborane/Watercomplex-Mediated Copolymerization of Methyl Methacrylate with Proteinaceous Materials and Proteins: a Review
Polymers 2010, 2, 575-595; doi:10.3390/polym2040575 OPEN ACCESS polymers ISSN 2073-4360 www.mdpi.com/journal/polymers Review Tri-n-Butylborane/WaterComplex-Mediated Copolymerization of Methyl Methacrylate with Proteinaceous Materials and Proteins: A Review Seiichiro Fujisawa 1,* and Yoshinori Kadoma 2 1 Meikai University School of Dentistry, Sakado, Saitama 350-0283, Japan 2 Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University, Kanda-surugadai, Chiyoda-ku, Tokyo 101-0062, Japan; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +81-049-279-2778; Fax: +81-492-876-657. Received: 23 August 2010; in revised form: 8 October 2010 / Accepted: 5 November 2010 / Published: 15 November 2010 Abstract: Previous studies of tri-n-butylborane–initiated graft copolymerization of methyl methacrylates with hydrated proteinous materials and proteins have focused on the number of grafted-poly (MMA) branches as well as the percent graft and graft efficiency. The number of branches in silk fibroin is 1.3, whereas the number in collagen, gelatin, ovalbumin and wool are 0.1, 0.04, 0.02 and 0.03, respectively. The number of grafted-PMMA branches in synthetic poly-L-peptides is approximately 10-fold less than that in gelatin, and decline, in the order poly-Ala > poly-Ser > poly-Pro > poly-Glu > poly-Lys. By contrast, poly-Gly, poly-Tyr and poly-Leu have no branches. The co-catalytic effect (the ratio of the number of polymer formed relative to that of control) of amino acids on tri-n-butylborane-initiated polymerization of MMA in the presence of water has been linearly correlated with their 2 ionization potential (IPkoopman); |Äå HOMO (Highest Occupied Molecular Orbital)| (r = 0.6, outliers: Cys and His); Äå HOMO = [åHOMOaqua − åHOMOvacuum] calculated using the semiempirical AM1 method. -
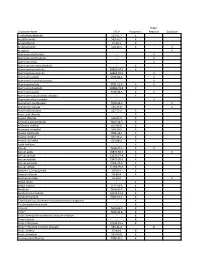
List of Major Physical Hazards
Water Substance Name CAS # Pyrophoric Reactive Explosive 2-ethylhexaldehyde 123-05-7 X Acetyl bromide 509-96-7 X Acetyl chloride 75-36-5 X Acetyl peroxide 110-22-5 X X Acetylene X Aluminum alkyl halides -- X Aluminum alkyl hydrides -- X Aluminum alkyls -- X Aluminum aminoborohydride -- X Aluminum borohydride 16962-07-5 X Aluminum borohydride 16962-07-5 X Aluminum carbide 1299-86-1 X Aluminum ferrosilcon powder -- X Aluminum hydride 7784-21-6 X X Aluminum phosphide 20859-73-8 X Aluminum powder 7429-90-5 X X Aluminum sesquibromide ethylate -- X Aluminum silicon powder -- X Ammonium perchlorate 7790-98-9 X Ammonium picrate 131-74-8 X Amyl trichlorosilane 107-72-2 X Anisic acid chloride -- X Anisoyl chloride 100-07-2 X Antimony pentachloride 7647-18-9 X Antimony, triethyl 617-85-6 X Antimony, trimethyl 594-10-5 X Arsenic trichloride 7784-34-1 X Arsenic, triethyl 617-75-4 X Arsenic, trimethyl 593-88-4 X Azido thallium -- X Barium 7440-39-3 X X Barium azide 18810-58-7 X X Barium carbide 12070-27-8 X Barium hydride 13477-09-3 X Barium peroxide 1304-29-6 X Barium sulfide 21109-95-5 X Benzene, 1,2-epoxyethyl 96-09-3 X Benzoyl chloride 98-88-4 X Benzoyl peroxide 94-36-0 X Benzyl silane -- X Benzyl sodium 1121-53-5 X Beryllium 7440-41-7 X Beryllium borohydride 30374-53-9 X Beryllium hydride 7787-52-2 X Bis(ethylamino) siloxenebis-cyclopentadienyl manganese -- X Bis-dimethylstibine oxide -- X Bismuth 7440-69-9 X Boron 7440-42-8 X Boron arsenotribromideboron chloride tetramer -- X Boron triazide -- X Boron tribromide 10294-33-4 X Boron trifluoride -

Hazardous Chemicals Handbook
Hazardous Chemicals Handbook Hazardous Chemicals Handbook Second edition Phillip Carson PhD MSc AMCT CChem FRSC FIOSH Head of Science Support Services, Unilever Research Laboratory, Port Sunlight, UK Clive Mumford BSc PhD DSc CEng MIChemE Consultant Chemical Engineer Oxford Amsterdam Boston London New York Paris San Diego San Francisco Singapore Sydney Tokyo Butterworth-Heinemann An imprint of Elsevier Science Linacre House, Jordan Hill, Oxford OX2 8DP 225 Wildwood Avenue, Woburn, MA 01801-2041 First published 1994 Second edition 2002 Copyright © 1994, 2002, Phillip Carson, Clive Mumford. All rights reserved The right of Phillip Carson and Clive Mumford to be identified as the authors of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988 No part of this publication may be reproduced in any material form (including photocopying or storing in any medium by electronic means and whether or not transiently or incidentally to some other use of this publication) without the written permission of the copyright holder except in accordance with the provisions of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London, England W1T 4LP. Applications for the copyright holder’s written permission to reproduce any part of this publication should be addressed to the publishers British Library Cataloguing in Publication Data A catalogue record for this book is available from the British Library Library of Congress Cataloguing -

Preparation and Lewis Acid Properties of Diboron Tetrafluoride and Related Compounds
AN ABSTRACT OF THE THESIS OF JOHN DONALD HANSEN for the Ph. D. in Inorganic Chemistry (Name) (Degree) (Major) Date thesis is presented February 1, 1965 Title PREPARATION AND LEWIS ACID PROPERTIES OF DIBORON TETRAFLUORIDE AND RELATED COMPOUNDS Abstract approved (Major professor) Attempts to synthesize diboron tetrafluoride by the reduction of boron trifluoride with alkali metal solutions in 1, 2 - dimethoxyethane proved to be unsuccessful. No diboron tetrahalide was found when boron trifluoride, antimony trifluoride, or boron trichloride was reacted with tetra -(dimethylamino) - diboron; although there appeared to be some exchange between chloride and amine groups. Diboron tetrafluoride was prepared by the treatment of diboron tetrachloride with antimony trifluoride. The diboron tetrachloride was produced by the acid hydrolysis of tetra -(dimethylamino) -diboron to hypoboric acid with subsequent dehydration and reaction with boron trichloride. From the results of.the formation of Lewis acid -base complexes and their acid displacement reactions the order of relative acid strength was found to be: hydrogen chloride, diboron tetrafluoride boron trifluoride <boron trichloride. Diboron tetrafluoride formed strong complexes with triethylamine, trimethylamine, 2 , 6- lutidine, N, N, N', N' -tetramethylethylenediamine, and 1, 2- dimethoxyethane. A weak complex occurred with benzoni- trile, whereas, no complexation was found with p- chlorobenzonitrile. Boron trifluoride displaced diboron tetrafluoride from its complexes with 1, 2- dimethoxyethane, trimethylamine, and triethylamine. No displacement occurred, under the conditions investigated, with the complexes of N, N, N', N'- tetramethylethylenediamine and benzonitrile. It was found that p- chlorobenzonitrile serves as an excellent Lewis base for the separation of boron trifluoride from hydrogen chloride and boron trichloride impurities by complex formation.