The Cis-Regulatory Effects of Modern Human-Specific Variants
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Genetic Determinants of Inter-Individual Variations in DNA Methylation
Genetic determinants of inter-individual variations in DNA methylation Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) der Fakultät für Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Julia Wimmer (geb. Wegner) aus Neubrandenburg im Jahr 2014 The present work was carried out at the Clinic and Polyclininc of Internal Medicine III at the University Hospital Regensburg from March 2010 to August 2014. Die vorliegende Arbeit entstand im Zeitraum von März 2010 bis August 2014 an der Klinik und Poliklinik für Innere Medizin III des Universitätsklinikums Regensburg. Das Promotionsgesuch wurde eingereicht am: 01. September 2014 Die Arbeit wurde angeleitet von: Prof. Dr. Michael Rehli Prüfungsausschuss: Vorsitzender: Prof. Dr. Herbert Tschochner Erstgutachter: Prof. Dr. Michael Rehli Zweitgutachter: Prof. Dr. Axel Imhof Drittprüfer: Prof. Dr. Gernot Längst Ersatzprüfer: PD Dr. Joachim Griesenbeck Unterschrift: ____________________________ Who seeks shall find. (Sophocles) Table of Contents TABLE OF CONTENTS .......................................................................................................................... I LIST OF FIGURES ................................................................................................................................ IV LIST OF TABLES ................................................................................................................................... V 1 INTRODUCTION .......................................................................................................................... -

A Neuro-Specific Hedgehog-Responsive Enhancer from Intron 1 of the Murine Laminin Alpha 1 Gene
A neuro-specific hedgehog-responsive enhancer from intron 1 of the murine laminin alpha 1 gene Thesis presented for the degree of PhD by Kalin Dimitrov Narov Department of Biomedical Science University of Sheffield September 2013 i Acknowledgements I thank my supervisors Anne-Gaëlle Borycki and Philip Ingham for all expert guidance, patience, and for the opportunity to study vertebrate development in their laboratories. I also thank my advisors Pen Rashbass and Vincent Cunliffe for the helpful advices and their critical analyses on my work, and our collaborator Norris Ray Dunn for advising me on mouse transgenics and providing me with mouse embryos. Many thanks also to Shantisree Rayagiri, Joseph B. Pickering, Claire Anderson, Ashish Maurya, Weixin Niah, Harriet Jackson, Raymond Lee, Xingang Wang, Yogavali Pooblan for helping me with text formatting and embryo harvesting, and for providing me with reagents and advices. I am also grateful to the whole D-floor community, as well as to Martin Zeidler’s and Marcelo Rivolta’s labs for letting me use their equipment. Last but not least, I thank my family for the constant support and encouragement, and especially my parents for nurturing in me the love to nature and knowledge. Therefore, I dedicate this work to the memory of my father. ii Abstract Laminin alpha 1 (LAMA1) is a major component of the earliest basement membranes in the mammalian embryo. Disruption of the murine Lama1 gene result in lethal failure of germ layer differentiation and extraembryonic membrane formation at gastrulation stages, while conditional deletion of Lama1 leads to aberrant organization of retinal neurons and vasculature, and defects in cerebellar glia and granule cell precursors later in development. -

Epigenetic & Transcriptomic Alterations Associated with Childhood
EPIGENETIC & TRANSCRIPTOMIC ALTERATIONS ASSOCIATED WITH CHILDHOOD MALTREATMENT, MAJOR DEPRESSIVE DISORDER, AND POST-TRAUMATIC STRESS DISORDER BY ANGELA CATHERINE BUSTAMANTE DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Neuroscience in the Graduate College of the University of Illinois at Urbana-Champaign, 2017 Urbana, Illinois Doctoral Committee: Associate Professor Monica Uddin, Chair Professor Karestan C. Koenen, Harvard University Associate Professor Alison M. Bell Professor Sandra Rodriguez-Zas ABSTRACT Childhood maltreatment increases the risk of developing major depressive disorder (MDD) and post-traumatic stress disorder (PTSD) later in life. Dysregulation of the hypothalamic-pituitary- adrenal (HPA) axis, a regulator of the body’s stress response, has been associated with exposure to childhood maltreatment, MDD, and PTSD. Additional work has shown that a history of childhood maltreatment, MDD, and PTSD is associated with altered DNA methylation levels within key HPA axis genes. Despite this well-known association, the molecular mechanism(s) contributing to the increased risk of developing mental illness, specifically MDD or PTSD, following exposure to childhood maltreatment remain poorly understood. Therefore, the goal of my dissertation is to further elucidate the biologic mechanisms contributing to the relationship among childhood maltreatment, MDD, and PTSD by focusing on DNA methylation and gene expression. Chapter 2 investigates whether childhood maltreatment and MDD have a joint or potentially interacting effect on NR3C1, the glucocorticoid receptor, promoter region DNA methylation using whole blood. A secondary analysis examines the potential functional effect on downstream gene expression levels. Chapter 3 examines whether blood-derived DNA methylation of FKBP5, another HPA axis gene, mediates the relationship between childhood maltreatment and MDD, and whether DNA methylation subsequently influences FKBP5 gene expression. -

Molecular Characterization of Occult Hepatitis B Virus Infection in Patients with End-Stage Liver Disease in Colombia
RESEARCH ARTICLE Molecular characterization of occult hepatitis B virus infection in patients with end-stage liver disease in Colombia Julio Cesar Rendon1, Fabian Cortes-Mancera2, Juan Carlos Restrepo-Gutierrez1,3, Sergio Hoyos1,3, Maria-Cristina Navas1* 1 Grupo de Gastrohepatologia, Facultad de Medicina, Universidad de Antioquia, UdeA, Medellin, Colombia, 2 Grupo de InvestigacioÂn e Innovacion BiomeÂdica GI2B, Facultad de Ciencias Exactas y Aplicadas, Instituto a1111111111 Tecnologico Metropolitano (ITM), Medellin, Colombia, 3 Unidad de Hepatologia y Trasplante Hepatico, a1111111111 Hospital Pablo Tobon Uribe, Medellin, Colombia a1111111111 a1111111111 * [email protected] a1111111111 Abstract OPEN ACCESS Background Citation: Rendon JC, Cortes-Mancera F, Restrepo- Hepatitis B virus (HBV) occult infection (OBI) is a risk factor to be taken into account in trans- Gutierrez JC, Hoyos S, Navas M-C (2017) fusion, hemodialysis and organ transplantation. The aim of this study was to identify and Molecular characterization of occult hepatitis B virus infection in patients with end-stage liver characterize at the molecular level OBI cases in patients with end-stage liver disease. disease in Colombia. PLoS ONE 12(7): e0180447. https://doi.org/10.1371/journal.pone.0180447 Methods Editor: Isabelle Chemin, Centre de Recherche en Sixty-six liver samples were obtained from patients with diagnosis of end-stage liver Cancerologie de Lyon, FRANCE disease submitted to liver transplantation in Medellin (North West, Colombia). Samples Received: February 1, 2017 obtained from patients who were negative for the surface antigen of HBV (n = 50) were Accepted: June 15, 2017 tested for viral DNA detection by nested PCR for ORFs S, C, and X and confirmed by South- Published: July 7, 2017 ern-Blot. -

PROGRAMMED FUNCTION of the TAMMAR MAMMARY GLAND; the Role of Milk Proteins in Gut Development
PROGRAMMED FUNCTION OF THE TAMMAR MAMMARY GLAND; The role of milk proteins in gut development by Sanjana Kuruppath Master of Science (MSc) Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Deakin University February, 2014 Acknowledgements Thank You Almighty! I thank my family, friends and colleagues for their support without which completion of this thesis would not have been possible. First and most of all, I would like to express my sincere and deepest gratitude to my PhD supervisor Professor Kevin R Nicholas, for his constant guidance and support. I would not have been in a position to write this if he had not selected me as a student, given an opportunity to come to Australia and pursue my career in the field of scientific research. I assure, I had the best supervisor for my PhD. Under him, I had the freedom to manage and execute my project with his continuous support and timely guidance because of which I gradually grew as a confident researcher. I was always surprised to see him welcoming his students with a smiling and encouraging face, anytime they knock at his door! I greatly appreciate his commitment and passion towards research and the magical ability to continuously and convincingly promote his students to grow independently. I also thank him, from the bottom of my heart, for all the support and courage he gave me in my personal life too. I thank my co-supervisors, Dr Christophe Lefevre and Dr Julie A Sharp. I thank Christophe for his advice and assistance, mainly in the field of bioinformatics. -
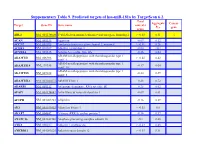
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Bayesian Infinite Mixture Models for Gene Clustering and Simultaneous
UNIVERSITY OF CINCINNATI Date: 22-Oct-2009 I, Johannes M Freudenberg , hereby submit this original work as part of the requirements for the degree of: Doctor of Philosophy in Biomedical Engineering It is entitled: Bayesian Infinite Mixture Models for Gene Clustering and Simultaneous Context Selection Using High-Throughput Gene Expression Data Student Signature: Johannes M Freudenberg This work and its defense approved by: Committee Chair: Mario Medvedovic, PhD Mario Medvedovic, PhD Bruce Aronow, PhD Bruce Aronow, PhD Michael Wagner, PhD Michael Wagner, PhD Jaroslaw Meller, PhD Jaroslaw Meller, PhD 11/18/2009 248 Bayesian Infinite Mixture Models for Gene Clustering and Simultaneous Context Selection Using High-Throughput Gene Expression Data A dissertation submitted to the Graduate School of the University of Cincinnati in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department Biomedical Engineering of the Colleges of Engineering and Medicine by Johannes M. Freudenberg M.S., 2004, Leipzig University, Leipzig, Germany (German Diplom degree) Committee Chair: Mario Medvedovic, Ph.D. Abstract Applying clustering algorithms to identify groups of co-expressed genes is an important step in the analysis of high-throughput genomics data in order to elucidate affected biological pathways and transcriptional regulatory mechanisms. As these data are becoming ever more abundant the integration with both, existing biological knowledge and other experimental data becomes as crucial as the ability to perform such analysis in a meaningful but virtually unsupervised fashion. Clustering analysis often relies on ad-hoc methods such as k-means or hierarchical clustering with Euclidean distance but model-based methods such as the Bayesian Infinite Mixtures approach have been shown to produce better, more reproducible results.