Pfizer-Biontech COVID-19 Vaccine VRBPAC Briefing Document
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Central Nervous System Dysfunction As the First Manifestation of Multiple
Bone Marrow Transplantation (2000) 25, 79–83 2000 Macmillan Publishers Ltd All rights reserved 0268–3369/00 $15.00 www.nature.com/bmt Central nervous system dysfunction as the first manifestation of multiple organ dysfunction syndrome in stem cell transplant patients B Gordon1, E Lyden2, J Lynch2, S Tarantolo3, ZS Pavletic3, M Bishop3, E Reed3, A Kessinger3 and W Haire3 Departments of 1Pediatrics, 2Preventive and Societal Medicine, and 3Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA Summary: Organ system dysfunction is a frequent complication of hematopoietic stem cell transplantation. Single and multior- Development of CNS dysfunction in the setting of hema- gan dysfunction may be due to the culmination of an uncon- topoietic stem cell transplant (HSCT) has been pre- trolled inflammatory response to some initial event, viously shown to predict for subsequent second organ mediated by an as yet incompletely understood interplay of dysfunction and death. In this paper, we describe the cytokines, complement, and the coagulation system. In the characteristics of this isolated CNS dysfunction, and its setting of acutely ill non-transplant patients, this has been relationship to multiple organ dysfunction syndrome termed the systemic inflammatory response syndrome.1 (MODS) after HSCT. Twenty-one of 186 patients This syndrome has been well described in patients with undergoing HSCT developed CNS dysfunction as their trauma or severe infection,1–3 but has only recently been first organ dysfunction a mean of 22.8 ± 0.9 days after recognized in patients undergoing HSCT. the start of the preparative regimen. Compared with The occurrence of single organ system dysfunction in 137 patients who developed no organ dysfunction, the post-transplant period has been shown to predict for patients presenting with CNS dysfunction were more subsequent development of multiple organ dysfunction and likely to have undergone allogeneic HSCT (P = 0.001) death. -

Main Outcomes of Discussion of WHO Consultation on Nucleic Acid
MAIN OUTCOMES OF DISCUSSION FROM WHO CONSULTATION ON NUCLEIC ACID VACCINES I. Knezevic, R. Sheets 21-23 Feb, 2018 Geneva, Switzerland CONTEXT OF DISCUSSION • WHO Consultation held to determine whether the existing DNA guidelines were due for revision or if they remained relevant with today’s status of nucleic acid vaccine development and maturity towards licensure (marketing authorization) • Presentation will cover alignment to existing guidelines • Current status of development of DNA and RNA vaccines, both prophylactic & therapeutic • Main outcomes of discussion • Next steps already underway & planned STATUS OF DEVELOPMENT OF NUCLEIC ACID VACCINES • First likely candidate to licensure could be a therapeutic DNA vaccine against Human Papilloma Viruses • Anticipated to be submitted for licensure in 3-5 years • Other DNA vaccines are likely to follow shortly thereafter, e.g., Zika prophylaxis • RNA vaccines – less clinical experience, but therapeutic RNAs anticipated in 2021/22 timeframe to be submitted for licensure • For priority pathogens in context of public health emergencies, several candidates under development (e.g., MERS-CoV, Marburg, Ebola) MAIN OUTCOMES - GENERAL • Regulators expressed need for updated DNA guideline for prophylaxis and therapy • Less need at present for RNA vaccines in guideline but need some basic PTC • Flexibility needed now until more experience gained for RNA vaccines that will come in next few years • Revisit need for a more specific guideline at appropriate time for RNA vaccines • Institutional Biosafety Committees are regulated by national jurisdictions & vary considerably • How can WHO assist to streamline or converge these review processes? Particularly, during Public Health Emergency – can anything be done beforehand? MAIN OUTCOMES - DEFINITIONS • Clear Definitions are needed • Proposed definition of DNA vaccine: • A DNA plasmid(s) into which the desired immunogen(s) is (are) encoded and prepared as purified plasmid preparations to be administered in vivo. -

Review of COVID-19 Vaccine Subtypes, Efficacy and Geographical Distributions Andre Ian Francis ,1 Saudah Ghany,1 Tia Gilkes,1 Srikanth Umakanthan2
Review Postgrad Med J: first published as 10.1136/postgradmedj-2021-140654 on 6 August 2021. Downloaded from Review of COVID-19 vaccine subtypes, efficacy and geographical distributions Andre Ian Francis ,1 Saudah Ghany,1 Tia Gilkes,1 Srikanth Umakanthan2 1Department of Clinical Medical ABSTRACT 2021, the Ad26.COV2.S, developed by Janssen Sciences, The Faculty of Medical As of 1 May 2021, there have been 152 661 445 (Johnson & Johnson) and Moderna on 30 April.4 Sciences, The University of the COVAX, coordinated by WHO, Gavi: The West Indies, St. Augustine, Covid-19 cases with 3 202 256 deaths globally. Trinidad and Tobago This pandemic led to the race to discover a vaccine Vaccine Alliance, the Coalition for Epidemic 2Pathology unit, Department to achieve herd immunity and curtail the damaging Preparedness Innovations (CEPI), acts as a of Paraclinical Sciences, The effects of Covid-19. This study aims to discuss the most programme that supports the development of University Of The West Indies, St. COVID-19 vaccine candidates and negotiates their Augustine, Trinidad and Tobago recent WHO-approved Covid-19 vaccine subtypes, their status and geographical scheduled updates as of 4 pricing to ensure low- and- middle- income countries have a fair shot at receiving vaccines.5 Correspondence to May 2021. The keywords “Covid-19, Vaccines, Pfizer, Mr. Andre Ian Francis, The BNT162b2, AstraZeneca, AZD1222, Moderna, mRNA- This article aims at discussing the most recent University of the West Indies, St. 1273, Janssen, Ad26.COV2.S” were typed into PubMed. WHO- approved COVID-19 vaccine subtypes, their Augustine, Trinidad and Tobago; Thirty Two relevant PubMed articles were included in status and geographical scheduled updates as of 4 andre. -

Common COVID-19 Myths and Misinformation
Common COVID-19 Myths and Misinformation Myth: The side effects of Myth: The COVID-19 Myth: If I’ve already had the COVID-19 vaccine are vaccine can affect COVID-19, I don’t need a dangerous. women’s fertility. vaccine. Myth: Researchers rushed the development of the Myth: Getting the COVID- Myth: The COVID-19 COVID-19 vaccine, so its 19 vaccine gives you vaccine can affect effectiveness and safety COVID-19. women’s fertility. cannot be trusted. Myth: The COVID-19 vaccine enters your cells and changes your DNA. Information gathered from the following website: https://www.hopkinsmedicine.org/health/conditions-and-diseases/coronavirus/covid-19-vaccines-myth-versus-fact https://www.muhealth.org/our-stories/covid-19-vaccine-myths-vs-facts FACT: People who have gotten sick with COVID-19 may still benefit from getting vaccinated. Due to the severe health risks associated with COVID-19 and the fact that re-infection with COVID-19 is possible, people may be advised to get a COVID-19 vaccine If I’ve already had even if they have been sick with COVID-19 before. There is not enough information currently available to COVID-19, I don’t say if or for how long people are protected from getting COVID-19 after they have had it (natural need a vaccine. immunity). Early evidence suggests natural immunity from COVID-19 may not last very long, but more studies are needed to better understand this. Several subjects in the Pfizer trial who were previously infected got vaccinated without ill effects. Some scientists believe the vaccine offers better protection for coronavirus than natural infection. -

Multiple Organ Dysfunction Syndrome Timothy G. Buchman, Ph.D., MD
Physiologic Failure: Multiple Organ Dysfunction Syndrome Timothy G. Buchman, Ph.D., M.D. Edison Professor of Surgery Professor of Anesthesiology and of Medicine Washington University School of Medicine St. Louis, Missouri 63110 [email protected] Acknowledgement: This work was supported, in part, by award GM48095 from the National Institutes of Health 2 Buchman Humans, like other life forms, can be viewed as thermodynamically open systems that continuously consume energy to maintain stability in the internal milieu in the face of ongoing environmental stress. In contrast to simple unicellular life forms such as bacteria, higher life forms must maintain stability not only in individual cells but also for the organism as a whole. To this end, a collection of physiologic systems evolved to process foodstuffs; to acquire oxygen and dispose of gaseous waste; to eliminate excess fluid and soluble toxins; and to perform other tasks. These systems—labeled respiratory, circulatory, digestive, neurological, and so on—share several features. • Physiological systems are spatially distributed. Food has to get from mouth to anus. Urine is made in the kidneys but has to exit the urethra. Blood may be pumped by the heart but has to reach the great toe. • Physiologic systems are generally spacefilling and structurally fractal. Each cell has demand for nutrients; each cell excretes waste. Information has to travel from the brain throughout the body. While not every physiologic system is self-similar at all levels of granularity, there is typically a nested architecture that facilitates function: microvillus to villus to intestinal mucosa; alveolus to alveolar unit to bronchial segment; capillary to arteriole to artery. -

V.5 3/18/2021 1 COVID-19 Vaccine FAQ Sheet
COVID-19 Vaccine FAQ Sheet (updated 3/18/2021) The AST has received queries from transplant professionals and the community regarding the COVID-19 vaccine. The following FAQ was developed to relay information on the current state of knowledge. This document is subject to change and will be updated frequently as new information or data becomes available. What kinds of vaccines are available or under development to prevent COVID-19? There are currently several vaccine candidates in use or under development. In the United States, the Government is supporting six separate vaccine candidates. Several other vaccines are also undergoing development outside of the United States government sponsorship and further information can be found here: NYTimes Coronavirus Vaccine Tracker: https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine- tracker.html Washington Post Vaccine Tracker: https://www.washingtonpost.com/graphics/2020/health/covid-vaccine-update- coronavirus/ v.5 3/18/2021 1 The types of vaccines are as follows (March 1, 2021) 1: Table 1: Vaccines Under Development or Available Through EUA Vaccine Type Compound Name [Sponsor] Clinical Notes Trial Phase mRNA mRNA-1273 [Moderna] Phase 3 Emergency use in U.S., E.U., other countries Approved in Canada BNT162b2 (Comirnaty) [Pfizer] Phase 2/3 Emergency use in U.S., E.U., other countries Also approved in Canada and other countries Replication- AZD1222 (Covishield) Phase 2/3 Emergency use defective [AstraZeneca] in U.K., India, adenoviral other countries vector (not U.S.) JNJ-78326735/Ad26.COV2.S -

Second Edition
COVID-19 Evidence Update COVID-19 Update from SAHMRI, Health Translation SA and the Commission on Excellence and Innovation in Health Updated 4 May 2020 – 2nd Edition “What is the prevalence, positive predictive value, negative predictive value, sensitivity and specificity of anosmia in the diagnosis of COVID-19?” Executive Summary There is widespread reporting of a potential link between anosmia (loss of smell) and ageusia (loss of taste) and SARS-COV-2 infection, as an early sign and with sudden onset predominantly without nasal obstruction. There are calls for anosmia and ageusia to be recognised as symptoms for COVID-19. Since the 1st edition of this briefing (25 March 2020), there has been a significant expansion of literature on this topic, including 3 systematic reviews. Predictive value: The reported prevalence of anosmia/hyposmia and ageusia/hypogeusia in SARS-COV-2 positive patients are in the order of 36-68% and 33-71% respectively. There are reports of anosmia as the first symptom in some patients. Estimates from one study for hyposmia and hypogeusia: • Positive likelihood ratios: 4.5 and 5.8 • Sensitivity: 46% and 62% • Specificity: 90% and 89% The US Centres for Disease Control and Prevention (CDC) has added new loss or taste or smell to its list of recognised symptoms for SARS-COV-2 infection. To date, the World Health Organization has not. Conclusion: There is sufficient evidence to warrant adding loss of taste and smell to the list of symptoms for COVID-19 and promoting this information to the public. Context • Early detection of COVID-19 is key to the ongoing management of the pandemic. -

Daily Unemployment Compensation Data
DISTRICT OF COLUMBIA DOES DISTRICT OF COLUMBIA DEPARTMENT OF DAILY UNEMPLOYMENT EMPLOYMENT SERVICES COMPENSATION DATA Preliminary numbers as of March 4, 2021.* Telephone Date Online Claims Daily Total Running Total Claims March 13, 2020 310 105 415 415 March 14, 2020 213 213 628 March 15, 2020 410 410 1,038 March 16, 2020 1,599 158 1,757 2,795 March 17, 2020 2,541 219 2,760 5,555 March 18, 2020 2,740 187 2,927 8,482 March 19, 2020 2,586 216 2,802 11,284 March 20, 2020 2,726 205 2,931 14,215 March 21, 2020 1,466 1,466 15,681 March 22, 2020 1,240 1,240 16,921 March 23, 2020 2,516 296 2,812 19,733 March 24, 2020 2,156 236 2.392 22,125 March 25, 2020 2,684 176 2,860 24,985 March 26, 2020 2,842 148 2,990 27,975 March 27, 2020 2,642 157 2,799 30,774 March 28, 2020 1,666 25 1,691 32,465 March 29, 2020 1,547 1,547 34,012 March 30, 2020 2,831 186 3,017 37,029 March, 31, 2020 2,878 186 3,064 40,093 April 1, 2020 2,569 186 2,765 42,858 April 2, 2020 2,499 150 2,649 45,507 April 3, 2020 2,071 300 2,371 47,878 April 4, 2020 1,067 14 1,081 48,959 April 5, 2020 1,020 1,020 49,979 April 6, 2020 2,098 155 2,253 52,232 April 7, 2020 1,642 143 1,715 54,017 April 8, 2020 1,486 142 1,628 55,645 *Recalculated and updated daily DISTRICT OF COLUMBIA Telephone DODISTRICT OF CEOLUMBIASDate Online Claims Daily Total Running Total DEPARTMENT OF DAILY UNEMPLOYMENTClaims EMPLOYMENT SERVICES April 9, 2020 1,604 111 1,715 57,360 April 10, 2020 COMPENSATION1,461 119 1,580 DATA58,940 April 11, 2020 763 14 777 59,717 April 12, 2020 698 698 60,415 April 13, 2020 1,499 104 -

Third Quarter 2020
March 31, 2020 Third Quarter 2020 Corporate update and financial results November 10, 2020 Forward-looking statements Various statements in this slide presentation concerning the future expectations of BioNTech, its plans and prospects, including the Company's views with respect to the potential for mRNA and other pipeline therapeutics; BioNTech's efforts to combat COVID-19; the collaborations between BioNTech and Pfizer and Fosun to develop a potential COVID-19 vaccine; our expectations regarding the potential characteristics of BNT162b2 in our continuing Phase 2/3 trial and/or in commercial use based on data observations to date; the expected timepoint for additional readouts on efficacy data of BNT162b2 in our Phase 2/3 trial; the nature of the clinical data for BNT162, BNT311 and our other product candidates, which is subject to ongoing peer review, regulatory review and market interpretation; the timing for submission of data for, or receipt of, any potential approval or Emergency Use Authorization with respect to our BNT162 program; the timing for submission of BNT162 manufacturing data to the FDA; the ability of BioNTech to supply the quantities of BNT162 to support clinical development and, if approved, market demand, including our production estimates for 2020 and 2021 and orders received to-date; the planned next steps in BioNTech's pipeline programs and specifically including, but not limited to, statements regarding plans to initiate clinical trials of BioNTech's product candidates and expectations for data announcements with -

Update on Approved and Candidate COVID-19 Vaccines in the EU
Update on approved and candidate COVID-19 vaccines in the EU Dr. Marco Cavaleri Head of Biological Health Threats and Vaccines Strategy, EMA An agency of the European Union Outline 1 EMA response to COVID-19 pandemic - milestones 2 COVID-19 vaccines approved in the EU 3 Benefits and risks of COVID-19 vaccines 4 Real world evidence on effectiveness 5 Studies in children 6 Vaccines under review by EMA 7 Adapting COVID-19 vaccines to variants 8 Additional information Classified as public by the European Medicines Agency EMA RESPONSE TO COVID-19 PANDEMIC Milestones in the fight against the pandemic Scientific & regulatory Rapid development & Transparency & mobilisation evaluation outreach Approval: Comirnaty, COVID-19 vaccine Moderna, COVID-19 vaccine AstraZeneca, COVID-19 vaccine Janssen Accelerated development & evaluation procedures WHO declares pandemic COVID-19 Experts’ Task Force Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec 2020 2021 1 Classified as public by the European Medicines Agency COVID-19 vaccines approved in the EU 4 vaccines authorised in the EU • Comirnaty and Moderna vaccines contain a molecule called messenger RNA (mRNA) with instructions for producing the spike protein from SARS-CoV-2, the virus that causes COVID-19 • The AstraZeneca and Janssen vaccine uses a non-replicating adenovirus as a carrier that has been modified to produce the spike protein from SARS-CoV-2. • The vaccines do not contain the SARS-CoV-2 virus causing COVID-19 itself and cannot cause the disease. Comirnaty COVID-19 Vaccine COVID-19 Vaccine COVID-19 Vaccine (BioNTech/Pfizer) Moderna AstraZeneca Janssen 21 Dec 6 Jan 29 Jan 11 Mar 2 Classified as public by the European Medicines Agency BENEFITS AND RISKS Efficacy of COVID-19 vaccines in trials All COVID-19 vaccines approved in the EU have a positive benefit-risk balance in prevention of COVID-19 disease. -

Unifying the Motor & Non-Motor Features of Parkinson's Disease
Unifying the Motor & Non-motor Features of Parkinson’s Disease Ali Shalash Professor of Neurology Chair of Ain Shams Movement Disorders Group Department of Neurology, Ain Shams Univeristy Cairo, Egypt AGENDA 1. Motor and non-motor Symptoms 2. Pathophysiology of Motor & NMS 3. Relation To Disease Course 4. NMS in motor subtypes 5. Motor & NMS fluctuation 6. Treatment of Motor & NMS: links Parkinson’s Disease • PD prevalence 1 % of populations older than 65 years (Abbas et al., 2018) • From 1990 to 2015, the number of with PD patients doubled to over 6 million, and double again to over 12 million by 2040 (Dorsey et al, 2018) • Aging populations, increasing longevity, decreasing smoking rates, and the by- products of industrialization MDS Clinical Diagnostic Criteria for PD Postuma et al 2015 MDS Clinical Diagnostic Criteria for PD Postuma et al 2015 Non-motor Symptoms of PD • Neuropsychiatric symptoms • Autonomic dysfunction • Depression up to 50-70 % • Drooling • Anxiety up to 60 % • Orthostatic hypotension 30–58% • Apathy 60 % • Urinary dysfunction • Psychosis up to 40% • Erectile dysfunction • Impulse control and related disorders • Gastrointestinal dysfunction 25–67% • Dementia, 24% to 40%. • Excessive sweating • Cognitive impairment 20-25% (other • Others than dementia mainly mild cognitive impairment) • Pain 30–85% • Fatigue 50% • Disorders of sleep and wakefulness • Olfactory dysfunction 90% • • Sleep fragmentation and insomnia Ophthalmologic dysfunction • Rapid eye movement sleep behavior disorder 65 % • Excessive daytime sleepiness Pathophysiology of Motor & NMS Neuropathology of PD: Motor Symptoms Two major pathologic processes: (a) premature selective loss of dopamine neurons: 30–70% cell loss ⇢ motor symptoms. (b) the accumulation of Lewy bodies, composed of α-synuclein Poewe, W. -
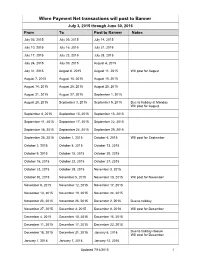
Transactions Posted to Pathway
When Payment Net transactions will post to Banner July 3, 2015 through June 30, 2016 From To Post to Banner Notes July 03, 2015 July 09, 2015 July 14, 2015 July 10, 2015 July 16, 2015 July 21, 2015 July 17, 2015 July 23, 2015 July 28, 2015 July 24, 2015 July 30, 2015 August 4, 2015 July 31, 2015 August 6, 2015 August 11, 2015 Will post for August August 7, 2015 August 13, 2015 August 18, 2015 August 14, 2015 August 20, 2015 August 25, 2015 August 21, 2015 August 27, 2015 September 1, 2015 August 28, 2015 September 3, 2015 September 9, 2015 Due to holiday at Monday Will post for August September 4, 2015 September 10, 2015 September 15, 2015 September 11, 2015 September 17, 2015 September 22, 2015 September 18, 2015 September 24, 2015 September 29, 2015 September 25, 2015 October 1, 2015 October 6, 2015 Will post for September October 2, 2015 October 8, 2015 October 13, 2015 October 9, 2015 October 15, 2015 October 20, 2015 October 16, 2015 October 22, 2015 October 27, 2015 October 23, 2015 October 29, 2015 November 3, 2015 October 30, 2015 November 5, 2015 November 10, 2015 Will post for November November 6, 2015 November 12, 2015 November 17, 2015 November 13, 2015 November 19, 2015 November 24, 2015 November 20, 2015 November 26, 2015 December 2, 2015 Due to holiday November 27, 2015 December 3, 2015 December 8, 2018 Will post for December December 4, 2015 December 10, 2015 December 15, 2015 December 11, 2015 December 17, 2015 December 22, 2015 December 18, 2015 December 31, 2015 January 6, 2016 Due to holiday closure Will post for December