An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Endogenous Transposable Element Tgm9 Is Suitable for Generating Knockout Mutants for Functional Analyses of Soybean Genes and Genetic Improvement in Soybean
RESEARCH ARTICLE The endogenous transposable element Tgm9 is suitable for generating knockout mutants for functional analyses of soybean genes and genetic improvement in soybean Devinder Sandhu1*, Jayadri Ghosh2, Callie Johnson3, Jordan Baumbach2, Eric Baumert3, Tyler Cina3, David Grant2,4, Reid G. Palmer2,4², Madan K. Bhattacharyya2* a1111111111 1 USDA-ARS, US Salinity Laboratory, Riverside, CA, United States of America, 2 Department of Agronomy, a1111111111 Iowa State University, Ames, IA, United States of America, 3 Department of Biology, University of Wisconsin- a1111111111 Stevens Point, Stevens Point, WI, United States of America, 4 USDA-ARS Corn Insects and Crop Genomics a1111111111 Research Unit, Ames, IA, United States of America a1111111111 ² Deceased. * [email protected] (DS); [email protected] (MKB) OPEN ACCESS Abstract Citation: Sandhu D, Ghosh J, Johnson C, In soybean, variegated flowers can be caused by somatic excision of the CACTA-type trans- Baumbach J, Baumert E, Cina T, et al. (2017) The posable element Tgm9 from Intron 2 of the DFR2 gene encoding dihydroflavonol-4-reduc- endogenous transposable element Tgm9 is suitable for generating knockout mutants for tase of the anthocyanin pigment biosynthetic pathway. DFR2 was mapped to the W4 locus, functional analyses of soybean genes and genetic where the allele containing Tgm9 was termed w4-m. In this study we have demonstrated improvement in soybean. PLoS ONE 12(8): that previously identified morphological mutants (three chlorophyll deficient mutants, one e0180732. https://doi.org/10.1371/journal. male sterile-female fertile mutant, and three partial female sterile mutants) were caused by pone.0180732 insertion of Tgm9 following its excision from DFR2. -

The Functional Readthrough Extension of Malate Dehydrogenase Reveals A
View metadata, citation and similar papersDownloaded at core.ac.uk from http://rsob.royalsocietypublishing.org/ on January 12, 2017 brought to you by CORE provided by MPG.PuRe The functional readthrough extension of malate dehydrogenase reveals a rsob.royalsocietypublishing.org modification of the genetic code Julia Hofhuis1, Fabian Schueren1, Christopher No¨tzel1,†, Thomas Lingner2, Jutta Ga¨rtner1, Olaf Jahn3 and Sven Thoms1 Research 1Department of Pediatrics and Adolescent Medicine, University Medical Center Go¨ttingen, University of Go¨ttingen, 37075 Go¨ttingen, Germany 2 Cite this article: Hofhuis J, Schueren F, Microarray and Deep Sequencing Core Facility, University Medical Center Go¨ttingen, University of Go¨ttingen, 37077 Go¨ttingen, Germany No¨tzel C, Lingner T, Ga¨rtner J, Jahn O, Thoms 3Proteomics Group, Max Planck Institute of Experimental Medicine, 37075 Go¨ttingen, Germany S. 2016 The functional readthrough extension ST, 0000-0003-3018-6363 of malate dehydrogenase reveals a modification of the genetic code. Open Biol. 6: Translational readthrough gives rise to C-terminally extended proteins, 160246. thereby providing the cell with new protein isoforms. These may have differ- http://dx.doi.org/10.1098/rsob.160246 ent properties from the parental proteins if the extensions contain functional domains. While for most genes amino acid incorporation at the stop codon is far lower than 0.1%, about 4% of malate dehydrogenase (MDH1) is physio- logically extended by translational readthrough and the actual ratio of Received: 26 August 2016 MDH1x (extended protein) to ‘normal’ MDH1 is dependent on the cell Accepted: 21 October 2016 type. In human cells, arginine and tryptophan are co-encoded by the MDH1x UGA stop codon. -

MDH1 and MPP7 Regulate Autophagy in Pancreatic Ductal Adenocarcinoma Maria New1,Tim Van Acker1, Jun-Ichi Sakamaki2, Ming Jiang3, Rebecca E
Published OnlineFirst February 14, 2019; DOI: 10.1158/0008-5472.CAN-18-2553 Cancer Molecular Cell Biology Research MDH1 and MPP7 Regulate Autophagy in Pancreatic Ductal Adenocarcinoma Maria New1,Tim Van Acker1, Jun-Ichi Sakamaki2, Ming Jiang3, Rebecca E. Saunders3, Jaclyn Long2, Victoria M.-Y. Wang4, Axel Behrens4, Joana Cerveira5, Padhmanand Sudhakar6,7,8, Tamas Korcsmaros6,7, Harold B.J. Jefferies1, Kevin M. Ryan2, Michael Howell3, and Sharon A. Tooze1 Abstract Pancreatic ductal adenocarcinoma (PDAC) is driven by genase 1), revealed their role in early stages of autophagy metabolic changes in pancreatic cells caused by oncogenic during autophagosome formation. MPP7 was involved in mutations and dysregulation of p53. PDAC cell lines and the activation of YAP1 (a transcriptional coactivator in the PDAC-derived xenografts grow as a result of altered meta- Hippo pathway), which in turn promoted autophagy, whereas bolic pathways, changes in stroma, and autophagy. Selective MDH1 was required for maintenance of the levels of the targeting and inhibition of one of these may open avenues essential autophagy initiator serine–threonine kinase ULK1, for the development of new therapeutic strategies. In this and increased in the activity upon induction of autophagy. study, we performed a genome-wide siRNA screen in a Our results provide a possible explanation for how autophagy PDAC cell line using endogenous autophagy as a readout is regulated by MPP7 and MDH1, which adds to our under- and identified several regulators of autophagy that were standing of autophagy regulation in PDAC. required for autophagy-dependent PDAC cell survival. Validation of two promising candidates, MPP7 (MAGUK Significance: This study identifies and characterizes MPP7 p55 subfamily member 7, a scaffolding protein involved in and MDH1 as novel regulators of autophagy, which is thought cell–cell contacts) and MDH1 (cytosolic Malate dehydro- to be responsible for pancreatic cancer cell survival. -

Table S1: Gene Symbol Full Gene Name Entrez Gene ID Refseq A2M
Table S1: Gene Symbol Full Gene Name Entrez Gene ID RefSeq A2M alpha-2-macroglobulin 2 NM_000014 ABHD15 abhydrolase domain containing 15 116236 NM_198147 ACADVL acyl-Coenzyme A dehydrogenase, very long chain 37 NM_000018 ACSS1 acyl-CoA synthetase short-chain family member 1 84532 NM_032501 ACY3 aspartoacylase (aminocyclase) 3 91703 NM_080658 ADAM33 ADAM metallopeptidase domain 33 80332 NM_153202 AFF2 AF4/FMR2 family, member 2 2334 NM_002025 ALX1 ALX homeobox 1 8092 NM_006982 ANGPTL4 angiopoietin-like 4 51129 NM_001039667 ANKRD20A3 ankyrin repeat domain 20 family, member A3 441425 NM_001012419 ANKRD45 ankyrin repeat domain 45 339416 NM_198493 ANXA1 annexin A1 301 NM_000700 ANXA5 annexin A5 308 NM_001154 APBB1IP amyloid beta (A4) precursor protein-binding, family B, member 1 interacting protein 54518 NM_019043 ARAP3 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 3 64411 NM_022481 ARF3 ADP-ribosylation factor 3 377 NM_001659 ARF5 ADP-ribosylation factor 5 381 NM_001662 ARHGAP1 Rho GTPase activating protein 1 392 NM_004308 ARHGAP6 Rho GTPase activating protein 6 395 NM_006125 ARHGDIA Rho GDP dissociation inhibitor (GDI) alpha 396 NM_004309 ARMC8 armadillo repeat containing 8 25852 NM_014154 ATP2A2 ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 488 NM_170665 ATP6AP2 ATPase, H+ transporting, lysosomal accessory protein 2 10159 NM_005765 ATP6V1B2 ATPase, H+ transporting, lysosomal 56/58kDa, V1 subunit B2 526 NM_001693 B3GNT8 UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 8 374907 NM_198540 B4GALNT1 beta-1,4-N-acetyl-galactosaminyl -
Download Validation Data
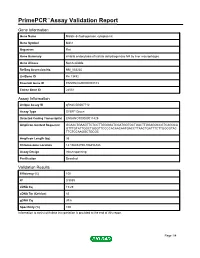
PrimePCR™Assay Validation Report Gene Information Gene Name Malate dehydrogenase, cytoplasmic Gene Symbol Mdh1 Organism Rat Gene Summary inhibits endocytosis of lactate dehydrogenase M4 by liver macrophages Gene Aliases Not Available RefSeq Accession No. NM_033235 UniGene ID Rn.13492 Ensembl Gene ID ENSRNOG00000008103 Entrez Gene ID 24551 Assay Information Unique Assay ID qRnoCID0007712 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000011429 Amplicon Context Sequence GCAACTGAAGTTCTCCTTGGGGATCGATGGTGCTGACTTGGAGGCCGTCAGGCA GTTTGTATTGGCTGGGTTCCCCACAACAATGACCTTAACTGATTTCTTGGCGTAC TTCTCCAAGGCTGCGC Amplicon Length (bp) 95 Chromosome Location 14:106454790-106456388 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9989 cDNA Cq 19.29 cDNA Tm (Celsius) 83 gDNA Cq 39.6 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Mdh1, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. Details for each transcript can be found on the Ensembl website at www.ensembl.org. -
CARM1-Expressing Ovarian Cancer Depends on the Histone Methyltransferase EZH2 Activity
ARTICLE DOI: 10.1038/s41467-018-03031-3 OPEN CARM1-expressing ovarian cancer depends on the histone methyltransferase EZH2 activity Sergey Karakashev1, Hengrui Zhu1, Shuai Wu1, Yuhki Yokoyama1, Benjamin G. Bitler1, Pyoung-Hwa Park1, Jeong-Heon Lee2, Andrew V. Kossenkov3, Krutika Satish Gaonkar4, Huihuang Yan4, Ronny Drapkin 5, Jose R. Conejo-Garcia6, David W. Speicher7, Tamas Ordog 2 & Rugang Zhang 1 CARM1 is an arginine methyltransferase that asymmetrically dimethylates protein substrates 1234567890():,; on arginine residues. CARM1 is often overexpressed in human cancers. However, clinically applicable cancer therapeutic strategies based on CARM1 expression remain to be explored. Here, we report that EZH2 inhibition is effective in CARM1-expressing epithelial ovarian cancer. Inhibition of EZH2 activity using a clinically applicable small molecule inhibitor sig- nificantly suppresses the growth of CARM1-expressing, but not CARM1-deficient, ovarian tumors in two xenograft models and improves the survival of mice bearing CARM1- expressing ovarian tumors. The observed selectivity correlates with reactivation of EZH2 target tumor suppressor genes in a CARM1-dependent manner. Mechanistically, CARM1 promotes EZH2-mediated silencing of EZH2/BAF155 target tumor suppressor genes by methylating BAF155, which leads to the displacement of BAF155 by EZH2. Together, these results indicate that pharmacological inhibition of EZH2 represents a novel therapeutic strategy for CARM1-expressing cancers. 1 Gene Expression and Regulation Program, The Wistar Institute, Philadelphia, PA 19104, USA. 2 Epigenomics Program, Center for Individualized Medicine, Mayo Clinic, Rochester, MN 55905, USA. 3 Center for Systems and Computational Biology, The Wistar Institute, Philadelphia, PA 19104, USA. 4 Division of Biostatistics and Informatics, Department of Health Science Research, Mayo Clinic, Rochester, MN 55905, USA. -

Wasin Vol. 9 N. 4 2558.Pmd
Asian Biomedicine Vol. 9 No. 4 August 2015; 455 - 471 DOI: 10.5372/1905-7415.0904.415 Original article Antiaging phenotype in skeletal muscle after endurance exercise is associated with the oxidative phosphorylation pathway Wasin Laohavinija, Apiwat Mutirangurab,c aFaculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand bDepartment of Anatomy, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand cCenter of Excellence in Molecular Genetics of Cancer and Human Diseases, Chulalongkorn University, Bangkok 10330, Thailand Background: Performing regular exercise may be beneficial to delay aging. During aging, numerous biochemical and molecular changes occur in cells, including increased DNA instability, epigenetic alterations, cell-signaling disruptions, decreased protein synthesis, reduced adenosine triphosphate (ATP) production capacity, and diminished oxidative phosphorylation. Objectives: To identify the types of exercise and the molecular mechanisms associated with antiaging phenotypes by comparing the profiles of gene expression in skeletal muscle after various types of exercise and aging. Methods: We used bioinformatics data from skeletal muscles reported in the Gene Expression Omnibus repository and used Connection Up- and Down-Regulation Expression Analysis of Microarrays to identify genes significant in antiaging. The significant genes were mapped to molecular pathways and reviewed for their molecular functions, and their associations with molecular and cellular phenotypes using the Database for Annotation, -
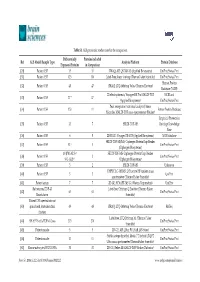
Table S1. ALS Proteomic Studies Used in the Comparison. Ref ALS Model/Sample Type Differentially Expressed Proteins Proteins
Table S1. ALS proteomic studies used in the comparison. Differentially Proteins included Ref ALS Model/Sample Type Analysis Platform Protein Database Expressed Proteins in Comparison [20] Patient CSF 35 35 iTRAQ; API QSTAR XL (Applied Biosystems) UniProt/Swiss-Prot [31] Patient CSF 123 110 Label-Free; linear ion trap (ThermoFisher Scientific) UniProt/Swiss-Prot Human Protein [32] Patient CSF 48 47 iTRAQ; LTQ-Orbitrap Velos (Thermo Electron) Database (NCBI) 2D electrophoresis; Voyager-DE Pro MALDI-TOF NCBI and [33] Patient CSF 17 * 17 (Applied Biosystems) UniProt/Swiss-Prot Peak recognition (statistical analysis); linear [34] Patient CSF 153 11 Entrez Protein Database Microflex MALDI-TOF mass spectrometer (Bruker) Empirical Proteomics [35] Patient CSF 33 7 SELDI-TOF-MS Ontology Knowledge Base [36] Patient CSF 6 5 2D-DIGE; Voyager DE-STR (Applied Biosystems) NCBI database SELDI-TOF-MS/MS; Ciphergen ProteinChip Reader [37] Patient CSF 52 † 3 UniProt/Swiss-Prot (Ciphergen Biosystems) 10 (PM-ALS) † SELDI-TOF-MS; Ciphergen ProteinChip Reader [38] Patient CSF 2 UniProt/Swiss-Prot 9 (L-ALS) † (Ciphergen Biosystems) [39] Patient CSF 3 2 SELDI-TOF-MS Unknown UHPLC LC-MS/MS; Q Exactive HF tandem mass [40] Patient CSF 3 3 UniProt spectrometer (Thermo Fisher Scientific) [41] Patient serum 7 7 2D-GE; SYNAPT-MS G1 (Waters Corporation) UniProt Rat neurons/TDP-43 Label-free; Orbitrap Q Exactive (Thermo Fisher [42] 63 63 UniProt/Swiss-Prot Knockdown Scientific) Patient CSF injected into rat [43] spinal cord /mitochondrial 49 48 iTRAQ; LTQ-Orbitrap Velos (Thermo Electron) RefSeq fraction Label-free; LTQ Orbitrap XL (Thermo Fisher [44] SH-SY5Y cells/TDP-43 Loss 273 270 UniProt/Swiss-Prot Scientific) [45] Patient muscle 5 5 2D-GE; API QStar PULSAR (AB-Sciex) UniProt/Swiss-Prot Stable-isotope dimethyl labels; 7 T hybrid LTQ FT [46] Patient muscle 11 11 UniProt/Swiss-Prot Ultra mass spectrometer (ThermoFisher Scientific) [47] Mouse astrocytes/SOD1 G93A 31 31 2D-GE; Reflex III MALDI-TOF (Bruker Daltonics) UniProt/Swiss-Prot Brain Sci. -

Molecular Analysis of Cytosolic and Mitochondrial Malate Dehydrogenases Isolated from Domestic Cats (Felis Catus)
Molecular analysis of cytosolic and mitochondrial malate dehydrogenases isolated from domestic cats (Felis catus) N. Sasaki1, M. Nakamura1 and S. Soeta2 1Laboratory of Veterinary Biochemistry, School of Veterinary Medicine, Nippon Veterinary and Life Science University, Tokyo, Japan 2Laboratory of Veterinary Anatomy, School of Veterinary Medicine, Nippon Veterinary and Life Science University, Tokyo, Japan Corresponding author: N. Sasaki E-mail: [email protected] Genet. Mol. Res. 13 (3): 6855-6864 (2014) Received July 8, 2013 Accepted January 4, 2014 Published August 29, 2014 DOI http://dx.doi.org/10.4238/2014.August.29.7 ABSTRACT. Malate dehydrogenase (MDH) plays crucial roles in energy and cellular metabolism. In this study, we describe the identification and characterization of cytosolic MDH (MDH1) and mitochondrial MDH (MDH2) in liver of domestic cat (Felis catus). To clone the feline full-length MDH genes, we performed rapid amplification of cDNA ends. The MDH1 gene encoded a protein of 334 amino acids and the MDH2 gene encoded a protein of 338 amino acids, containing a 24-amino acid mitochondrial target sequence. The feline MDH1 and MDH2 proteins shared, respectively, 98.8-93.7 and 96.7-94.4% homology with dog, giant panda, horse, cow, pig, human, mouse, and rat. The feline MDHs had a highly conserved active motif, which contained important residues for catalysis and coenzyme binding. The putatively acetylated lysine residues that regulate MDH activity were also conserved at K118, K121, and K298 in MDH1, and K185, K301, K307, and K314 in MDH2. Both MDH1 and MDH2 Genetics and Molecular Research 13 (3): 6855-6864 (2014) ©FUNPEC-RP www.funpecrp.com.br N. -

Tobacco Smoking Induces Metabolic Reprogramming of Renal Cell Carcinoma
The Journal of Clinical Investigation CLINICAL MEDICINE Tobacco smoking induces metabolic reprogramming of renal cell carcinoma James Reigle,1,2,3 Dina Secic,1,4 Jacek Biesiada,5 Collin Wetzel,1,6 Behrouz Shamsaei,5 Johnson Chu,1 Yuanwei Zang,1,7 Xiang Zhang,8 Nicholas J. Talbot,1 Megan E. Bischoff,1 Yongzhen Zhang,1,7 Charuhas V. Thakar,9,10 Krishnanath Gaitonde,9,11 Abhinav Sidana,11 Hai Bui,10 John T. Cunningham,1 Qing Zhang,12 Laura S. Schmidt,13,14 W. Marston Linehan,13 Mario Medvedovic,2,5 David R. Plas,1 Julio A. Landero Figueroa,4,15 Jarek Meller,2,3,5,15,16 and Maria F. Czyzyk-Krzeska1,10,15 1Department of Cancer Biology and 2Department of Biomedical Informatics, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA. 3Division of Biomedical Informatics, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio, USA. 4Agilent Metallomics Center of the Americas, Department of Chemistry, University of Cincinnati College of Arts and Science, Cincinnati, Ohio, USA. 5Division of Biostatistics and Bioinformatics, Department of Environmental and Public Health Sciences, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA. 6Rieveschl Laboratories for Mass Spectrometry, Department of Chemistry, University of Cincinnati College of Arts and Science, Cincinnati, Ohio, USA. 7Department of Urology, Qilu Hospital, Shandong University, Jinan, China. 8Division of Environmental Genetics and Molecular Toxicology, Department of Environmental and Public Health Sciences, and 9Division of Nephrology, Department of Medicine, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA. 10Cincinnati Veteran Affairs Medical Center, Department of Veterans Affairs, Cincinnati, Ohio, USA. 11Division of Urology, Department of Surgery, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA. -

Anti-CARM1 / PRMT4 Antibody (ARG53996)
Product datasheet [email protected] ARG53996 Package: 100 μl anti-CARM1 / PRMT4 antibody Store at: -20°C Summary Product Description Mouse Monoclonal antibody recognizes CARM1 / PRMT4 Tested Reactivity Hu, Ms Tested Application IP, WB Host Mouse Clonality Monoclonal Isotype IgG1 Target Name CARM1 / PRMT4 Species Human Immunogen E.coli derived recombinant human PRMT4/CARM1 protein fragments. Conjugation Un-conjugated Alternate Names Protein arginine N-methyltransferase 4; EC 2.1.1.125; PRMT4; EC 2.1.1.-; Coactivator-associated arginine methyltransferase 1; Histone-arginine methyltransferase CARM1 Application Instructions Application table Application Dilution IP Assay-dependent WB 1:200 - 1:500 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Properties Form Liquid Purification Affinity purified Buffer 100 mM Tris (pH 7.4), 150 mM NaCl, 0.1 mg/ml BSA, 50% Glycerol and 0.2% Sodium azide. Preservative 0.2% Sodium azide Stabilizer 0.1 mg/ml BSA, 50% Glycerol Concentration 1 mg/ml Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/3 Bioinformation Database links GeneID: 10498 Human GeneID: 59035 Mouse Swiss-port # Q86X55 Human Swiss-port # Q9WVG6 Mouse Gene Symbol CARM1 Gene Full Name coactivator-associated arginine methyltransferase 1 Background Methylates (mono- and asymmetric dimethylation) the guanidino nitrogens of arginyl residues in several proteins involved in DNA packaging, transcription regulation, pre-mRNA splicing, and mRNA stability. -
A Hypermethylation Strategy Utilized by Enhancer-Bound CARM1 to Promote Estrogen Receptor Α-Dependent Transcriptional Activatio
A hypermethylation strategy utilized by enhancer-bound CARM1 to promote estrogen receptor -dependent transcriptional activation and breast carcinogenesis Bing-ling Peng1,3, Wen-juan Li1,3, Jian-cheng Ding1,3, Yao-hui He1,3, Ting Ran1,3, Bing-lan Xie1, Zi-rui Wang1, Hai-feng Shen1, Rong-quan Xiao1, Wei-wei Gao1, Tian-yi Ye1, Xiang Gao1, Wen Liu1,2,* 1Fujian Provincial Key Laboratory of Innovative Drug Target Research, School of Pharmaceutical Sciences, Xiamen University, Xiang’an South Road, Xiamen, Fujian 361102, China 2State Key Laboratory of Cellular Stress Biology, School of Pharmaceutical Sciences, Xiamen University, Xiang’an South Road, Xiamen, Fujian 361102, China 3 These authors contributed equally *Correspondence: [email protected] ABSTRACT While protein arginine methyltransferases (PRMTs) and PRMT-catalyzed protein methylation have been well-known to be involved in a myriad of biological processes, their functions and the underlying molecular mechanisms in cancers, particularly in estrogen receptor alpha (ER)-positive breast cancers, remain incompletely understood. Here we focused on investigating PRMT4 (also called coactivator associated arginine methyltransferase 1, CARM1) in ER-positive breast cancers due to its high expression and the associated poor prognosis. Methods: ChIP-seq and RNA-seq were employed to identify the chromatin-binding landscape and transcriptional targets of CARM1, respectively, in the presence of estrogen in ER-positive MCF7 breast cancer cells. High-resolution mass spectrometry analysis of enriched peptides from anti- monomethyl- and anti-asymmetric dimethyl-arginine antibodies in SILAC labeled wild-type and CARM1 knockout cells were performed to globally map CARM1 methylation substrates. Cell viability was measured by MTS and colony formation assay, and cell cycle was measured by FACS analysis.