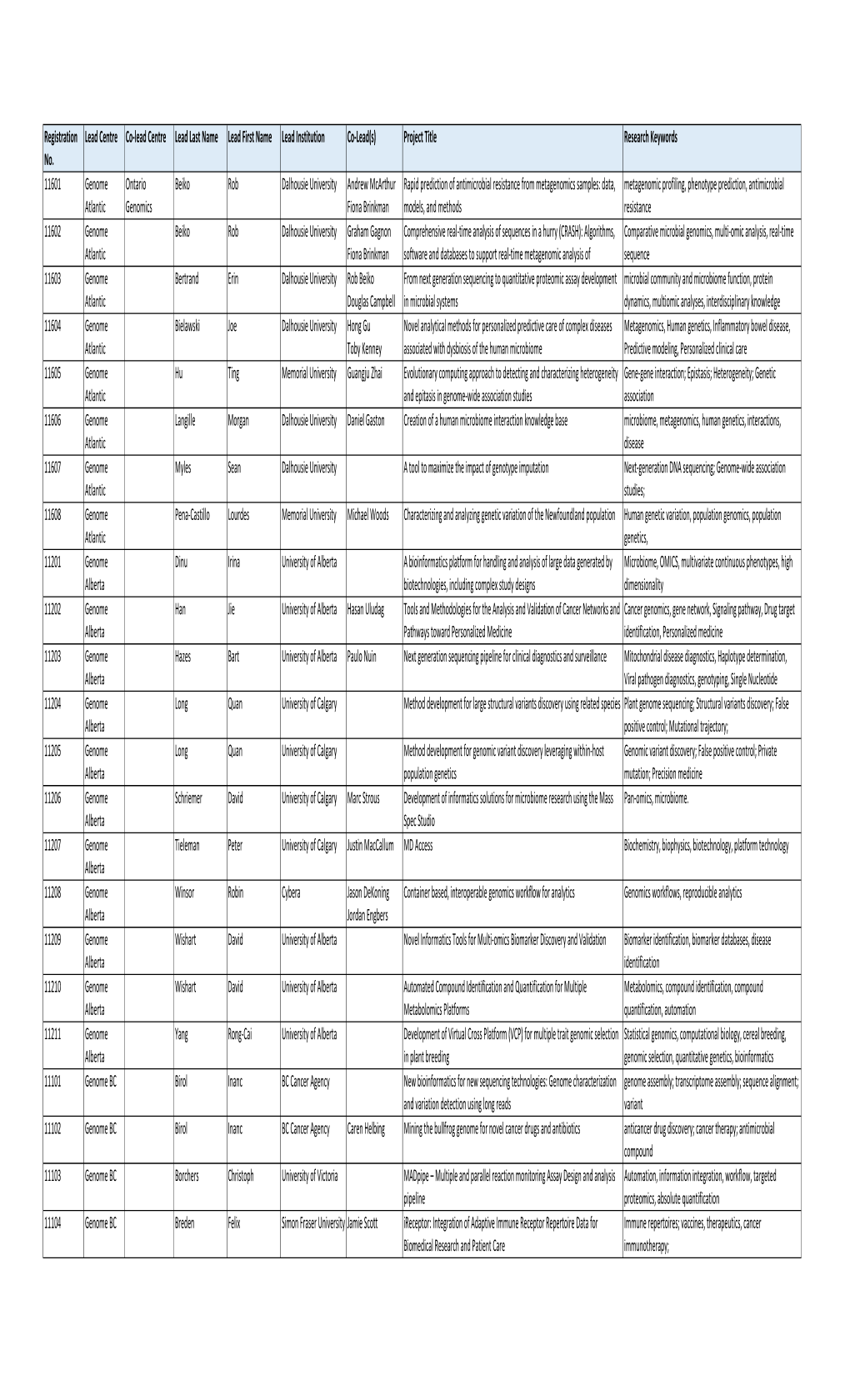
Summary of Registrations for Web 2 8 2016.Xlsx
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic, Epidemiological and Biological Analysis of Interleukin-10
Genes and Immunity (2009) 10, 174–180 & 2009 Macmillan Publishers Limited All rights reserved 1466-4879/09 $32.00 www.nature.com/gene ORIGINAL ARTICLE Genetic, epidemiological and biological analysis of interleukin-10 promoter single-nucleotide polymorphisms suggests a definitive role for À819C/T in leprosy susceptibility AC Pereira1,5, VN Brito-de-Souza1,5, CC Cardoso2, IMF Dias-Baptista1, FPC Parelli1,3, J Venturini1,3, FR Villani-Moreno1, AG Pacheco4 and MO Moraes2 1Instituto Lauro de Souza Lima, Bauru, Sa˜o Paulo, Brazil; 2Laborato´rio de Hansenı´ase, Departamento de Micobacterioses, Instituto Oswaldo Cruz, Rio de Janeiro, Brazil; 3Tropical Diseases Area, Botucatu Medical School, Sao Paulo State University, UNESP, Botucatu, Sa˜o Paulo, Brazil and 4Departamento de Epidemiologia e Me´todos Quantitativos em Sau´de (DEMQS), Escola Nacional de Sau´de Pu´blica (ENSP)/PROCC, FIOCRUZ, Rio de Janeiro, Brazil Leprosy is a complex infectious disease influenced by genetic and environmental factors. The genetic contributing factors are considered heterogeneous and several genes have been consistently associated with susceptibility like PARK2, tumor necrosis factor (TNF), lymphotoxin-a (LTA) and vitamin-D receptor (VDR). Here, we combined a case–control study (374 patients and 380 controls), with meta-analysis (5 studies; 2702 individuals) and biological study to test the epidemiological and physiological relevance of the interleukin-10 (IL-10) genetic markers in leprosy. We observed that the À819T allele is associated with leprosy susceptibility either in the case–control or in the meta-analysis studies. Haplotypes combining promoter single-nucleotide polymorphisms also implicated a haplotype carrying the À819T allele in leprosy susceptibility (odds ratio (OR) ¼ 1.40; P ¼ 0.01). -

PLINK: a Toolset for Whole Genome Association and Population-Based Linkage Analyses
1 PLINK: a toolset for whole genome association and population-based linkage analyses Shaun Purcell1,2, Benjamin Neale1,2,3, Kathe Todd-Brown1, Lori Thomas1, Manuel A R Ferreira1, David Bender1,2, Julian Maller1,2, Paul I W de Bakker1,2, Mark J Daly1,2, Pak C Sham4 1 Center for Human Genetic Research, MGH, Boston, USA. 2 Broad Institute of Harvard and MIT, Cambridge, USA. 3 Institute of Psychiatry, University of London, London, UK. 4 Genome Research Center, University of Hong Kong, Pokfulam, Hong Kong. Correspondence: Shaun Purcell, Rm 6.254, CPZ-N, 185 Cambridge Street, Boston, MA, 02114, USA; Tel: 617-726-7642; Fax: 617-726-0830; [email protected] Keywords : Whole genome association studies, single nucleotide polymorphisms, identity-by-state, identity-by-descent, linkage analysis, computer software Abstract Whole-genome association studies (WGAS) bring new computational as well as analytic challenges to researchers. Many existing genetic analysis tools are not designed to handle such large datasets in a convenient manner and do not necessarily exploit the new opportunities that whole-genome data bring. To address these issues, we developed PLINK, an open source C/C++ WGAS toolset. Large datasets, comprising hundreds of thousands of markers geno- typed for thousands of individuals, can be rapidly manipulated and analyzed in their entirety. As well as providing tools to make the basic analytic steps computationally efficient, PLINK also supports some novel approaches to whole-genome data, that take advantage of whole-genome coverage. We in- troduce PLINK and describe the five main domains of function: data man- agement, summary statistics, population stratification, association analysis and identity-by-descent estimation. -

Basics in Genetics Analysis
Genetics and Diseases Basics in Genetics Analysis Heping Zhang Environment 9/24/2007 Dr. Doug Brutlag Lecture Syllabus “central paradigm” //www.s-star.org/ 2 Diseases Progression Known and Probable Risk Factors z Being a woman How does the Breast Cancer grows and spread? z Getting older z Having a personal history of BC or ovarian cancer z Having a family history of breast cancer z Having a previous biopsy showing carcinoma in situ z Having your first period before age 12 z Starting menopause after age 55 A malignant Malignant Cancer cells z Never having children tumor within tumors spread z Having your first child after age 30 the breast suppress z Having a mutation in the BRCA1 or BRCA2 genes normal ones z Drinking more than 1 alcoholic drink per day z Being overweight after menopause or gaining weight as an adult. 9/24/2007 3 9/24/2007 4 Genetic Epidemiology Terminology • How is a disease transmitted in families? ( Marker: a known DNA sequence that can be identified by a Inheritance patterns). simple assay e.g., D1S80, D4S43, D16S126 • What is the recurrence Allele: a viable DNA coding that occupies a given locus risk for relatives? (position) on a chromosome e.g., A, a; • Mendelian disorders Genotype: the observed alleles at a genetic locus for an • Autosomal or X-linked individual • Dominant or recessive e.g., AA, Aa, aa; 0 Homozygous: AA, aa 0 Heterozygous: Aa Phenotype: the expression of a particular genotype 0 Continuous: blood pressure 0 Dichotomous: Cancer, Hypertension 9/24/2007 Duke University Center of Human Genetics 5 9/24/2007 6 1 Mendel’s Laws DNA Polymorphism and Human Variation First Law Segregation of Characteristics: the sex cell of a plant or animal may contain one factor (allele) for different traits but not both factors needed to express the traits. -

Genetic Association Tests for Binary Traits with An
GENETIC ASSOCIATION TESTS FOR BINARY TRAITS WITH AN APPLICATION by SULGI KIM Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Dissertation Advisor: Dr. Robert C. Elston Department of Epidemiology and Biostatistics CASE WESTER RESERVE UNIVERSITY August, 2009 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approved the dissertation of Sulgi Kim candidate for the Ph. D. degree*. (Signed) Robert C. Elston, Ph.D Department of Epidemiology and Biostatistics Chair of the committee Xiaofeng Zhu, Ph.D Department of Epidemiology and Biostatistics Courtney Gray-McGuire, Ph.D Department of Epidemiology and Biostatistics Jill S. Barnholtz-Sloan, Ph.D Case Comprehensive Cancer Center June 8, 2009 *We also certify that written approval has been obtained for any proprietary material therein. ii Table of Contents List of Tables .................................................................................................................... iv List of Figures .....................................................................................................................v Acknowledgements .......................................................................................................... vi Abstract ...............................................................................................................................1 Chapter 1. Introduction.....................................................................................................3 Chapter 2. Association Tests -

Design and Analysis of Genetic Association Studies
ection S ON Design and Analysis of Statistical Genetic Association G enetics Studies Hemant K Tiwari, Ph.D. Professor & Head Section on Statistical Genetics Department of Biostatistics School of Public Health Association Analysis • Linkage Analysis used to be the first step in gene mapping process • Closely located SNPs to disease locus may co- segregate due to linkage disequilibrium i.e. allelic association due to linkage. • The allelic association forms the theoretical basis for association mapping Linkage vs. Association • Linkage analysis is based on pedigree data (within family) • Association analysis is based on population data (across families) • Linkage analyses rely on recombination events • Association analyses rely on linkage disequilibrium • The statistic in linkage analysis is the count of the number of recombinants and non-recombinants • The statistical method for association analysis is “statistical correlation” between Allele at a locus with the trait Linkage Disequilibrium • Over time, meiotic events and ensuing recombination between loci should return alleles to equilibrium. • But, marker alleles initially close (genetically linked) to the disease allele will generally remain nearby for longer periods of time due to reduced recombination. • This is disequilibrium due to linkage, or “linkage disequilibrium” (LD). Linkage Disequilibrium (LD) • Chromosomes are mosaics Ancestor • Tightly linked markers Present-day – Alleles associated – Reflect ancestral haplotypes • Shaped by – Recombination history – Mutation, Drift Tishkoff -

Genome-Wide Association Studies: Understanding the Genetics of Common Disease the Academy of Medical Sciences | FORUM
The Academy of Medical Sciences | FORUM Genome-wide association studies: understanding the genetics of common disease Symposium report July 2009 The Academy of Medical Sciences The Academy of Medical Sciences promotes advances in medical science and campaigns to ensure these are converted into healthcare benefits for society. Our Fellows are the UK’s leading medical scientists from hospitals and general practice, academia, industry and the public service. The Academy seeks to play a pivotal role in determining the future of medical science in the UK, and the benefits that society will enjoy in years to come. We champion the UK’s strengths in medical science, promote careers and capacity building, encourage the implementation of new ideas and solutions – often through novel partnerships – and help to remove barriers to progress. The Academy’s FORUM with industry The Academy’s FORUM is an active network of scientists from industry and academia, with representation spanning the pharmaceutical, biotechnology and other health product sectors, as well as trade associations, Research Councils and other major charitable research funders. Through promoting interaction between these groups, the FORUM aims to take forward national discussions on scientific opportunities, technology trends and the associated strategic choices for healthcare and other life-science sectors. The FORUM builds on what is already distinctive about the Academy: its impartiality and independence, its focus on research excellence across the spectrum of clinical and basic sciences and its commitment to interdisciplinary working. Acknowledgements This report provides a summary of the discussion at the FORUM symposium on ‘Genome-wide association studies’ held in October 2008. -

Study of Genetic Association
Statistical Genomics and Bioinformatics Workshop 8/16/2013 Statistical Genomics and Bioinformatics Workshop: Genetic Association and RNA-Seq Studies Population Genetics and Genome‐wide Genetic Association Studies (GWAS) Brooke L. Fridley, PhD University of Kansas Medical Center 1 Study of Genetic Association Cases Controls Genetic association studies look at the frequency of genetic changes in a group of cases and controls to try to determine whether specific changes are associated with disease. 2 1 Statistical Genomics and Bioinformatics Workshop 8/16/2013 Genetic Analysis Strategies Linkage effect GWAS Association size Rare Variant Analysis allele frequency Ardlie, Kruglyak & Seielstad (2002) Nature Genetics Reviews Zondervan & Cardon (2004) Nature Genetics Reviews 3 Genetics of Complex Traits • Multiple genes / variants – Common and rare variants – Interactions, Haplotypes, Pathways • Environment – Gene‐Environment interaction 4 2 Statistical Genomics and Bioinformatics Workshop 8/16/2013 In reality, much more complex! 5 NIHGRI GWAS Catalog (8/11/2013) http://www.genome.gov/gwastudies/ 6 3 Statistical Genomics and Bioinformatics Workshop 8/16/2013 Population Genetics 7 Recombination A1 B1 D1 Before meioses A2 B2 D2 A1 B1 D2 Crossovers occur during meioses A2 B2 D1 D2 A1 B1 After recombination A2 B2 D1 8 4 Statistical Genomics and Bioinformatics Workshop 8/16/2013 Linkage Disequilibrium (LD) • Particular alleles at neighboring loci tend to be co-inherited. • For tightly linked loci, this co-inheritance might lead to associations between -

Genetic Association Analysis of SARS-Cov-2 Infection in 455,838 UK Biobank Participants
medRxiv preprint doi: https://doi.org/10.1101/2020.10.28.20221804; this version posted November 3, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-ND 4.0 International license . 1 Genetic association analysis of SARS-CoV-2 infection in 455,838 UK Biobank participants 2 J. A. Kosmicki†, J. E. Horowitz†, N. Banerjee, R. Lanche, A. Marcketta, E. Maxwell, Xiaodong 3 Bai, D. Sun, J. Backman, D. Sharma, C. O’Dushlaine, A. Yadav, A. J. Mansfield, A. Li, J. 4 Mbatchou, K. Watanabe, L. Gurski, S. McCarthy, A. Locke, S. Khalid, O. Chazara, Y. Huang, E. 5 Kvikstad, A. Nadkar, A. O’Neill, P. Nioi, M. M. Parker, S. Petrovski, H. Runz, J. D. Szustakowski, 6 Q. Wang, Regeneron Genetics Center*, UKB Exome Sequencing Consortium*, M. Jones, S. 7 Balasubramanian, W. Salerno, A. Shuldiner, J. Marchini, J. Overton, L. Habegger, M. N. Cantor, 8 J. Reid, A. Baras‡, G. R. Abecasis‡, M. A. Ferreira‡ 9 10 From: 11 Regeneron Genetics Center, 777 Old Saw Mill River Rd., Tarrytown, NY 10591, USA (JK, JEH, 12 NB, RL, AM, EM, XB, DS, JB, DSh, CO’D, AY, AJM, AL, JM, KW, LG, SM, AL, SK, MJ, SB, 13 WS, AS, JM, JO, LH, MNC, JR, AB, GRA, MAF) 14 Centre for Genomics Research, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, 15 Cambridge, UK (OC, AO’N, SP, QW) 16 Biogen, 300 Binney St, Cambridge, MA 02142, USA (YH, HR) 17 Alnylam Pharmaceuticals, 675 West Kendall St, Cambridge, MA 02142, USA (MMP, PN). -

Characterization of Quantitative Traits Using Association Genetics
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2010 Characterization of quantitative traits using association genetics tetraploid and genetic linkage mapping in diploid cotton (Gossypium spp.) Ashok Badigannavar Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Recommended Citation Badigannavar, Ashok, "Characterization of quantitative traits using association genetics tetraploid and genetic linkage mapping in diploid cotton (Gossypium spp.)" (2010). LSU Doctoral Dissertations. 2207. https://digitalcommons.lsu.edu/gradschool_dissertations/2207 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. CHARACTERIZATION OF QUANTITATIVE TRAITS USING ASSOCIATION GENETICS IN TETRAPLOID AND GENETIC LINKAGE MAPPING IN DIPLOID COTTON (GOSSYPIUM SPP.) A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The School of Plant, Environmental, and Soil Sciences By Ashok Badigannavar BSc (Agri), University of Agricultural Sciences, Dharwad, India, 1997 MSc (Agri), University of Agricultural Sciences, Dharwad, India, 1999 May 2010 This dissertation is dedicated in memory of my late father….. ii ACKNOWLEDGEMENTS I would like to express my sincere gratitude to my major professor Dr.Gerald O Myers for his valuable guidance and advice throughout my dissertation. The freedom he has provided during the course of study is commendable. -
![[Thesis Title Goes Here]](https://docslib.b-cdn.net/cover/8640/thesis-title-goes-here-2688640.webp)
[Thesis Title Goes Here]
RARE AND COMMON GENETIC VARIANT ASSOCIATIONS WITH QUANTITATIVE HUMAN PHENOTYPES A Dissertation Presented to The Academic Faculty by Jing Zhao In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the School of Biology Georgia Institute of Technology August 2015 COPYRIGHT © 2015 BY JING ZHAO RARE AND COMMON GENETIC VARIANT ASSOCIATIONS WITH QUANTITATIVE HUMAN PHENOTYPES Approved by: Dr. Greg Gibson, Advisor Dr. Joseph Lachance School of Biology School of Biology Georgia Institute of Technology Georgia Institute of Technology Dr. Fredrik Vannberg Dr. Eva Lee School of Biology School of Industrial and Systems Georgia Institute of Technology Engineering Georgia Institute of Technology Dr. Patrick McGrath School of Biology Georgia Institute of Technology Date Approved: June 25, 2015 To my family and friends ACKNOWLEDGEMENTS The path to a PhD is full of laughter and tears. During the long journey of pursuing the degree of PhD, many people have provided me help and encouragement. Among those people, thesis advisor plays a very important role in my success of graduate study and research thesis. I always feel grateful to have Dr. Greg Gibson as my PhD advisor for almost five years. I remember a lot of moments when I was frustrated with the disappointing research results, Dr. Gibson encouraged me and helped me find the right direction to solve the problems. His encouragement and support has boosted my strong belief to overcome the challenges and keep my enthusiasm for science. What is more important, his attitude about science will have long-term influence on my future career. I appreciate the guidance and advice from my thesis committee members: Dr. -

Geographic Confounding in Genome-Wide Association Studies
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.18.435971; this version posted March 18, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Geographic Confounding in Genome-Wide Association Studies Abdel Abdellaoui1, Karin J.H. Verweij1, Michel G. Nivard2 1Department of Psychiatry, Amsterdam UMC, University of Amsterdam, Amsterdam, the Netherlands 2Department of Biological Psychology, VU University, Amsterdam, the Netherlands Correspondence: [email protected]; [email protected]; [email protected] Abstract Gene-environment correlations can bias associations between genetic variants and complex traits in genome-wide association studies (GWASs). Here, we control for geographic sources of gene- environment correlation in GWASs on 56 complex traits (N=69,772–271,457). Controlling for geographic region significantly decreases heritability signals for SES-related traits, most strongly for educational attainment and income, indicating that socio-economic differences between regions induce gene-environment correlations that become part of the polygenic signal. For most other complex traits investigated, genetic correlations with educational attainment and income are significantly reduced, most significantly for traits related to BMI, sedentary behavior, and substance use. Controlling for current address has greater impact on the polygenic signal than birth place, suggesting both active and passive sources of gene-environment correlations. Our results show that societal sources of social stratification that extend beyond families introduce regional-level gene-environment correlations that affect GWAS results. -

A Tutorial on Statistical Methods for Population Association Studies
FOCUS ON STATISTICAL ANALYSISREVIEWS A tutorial on statistical methods for population association studies David J. Balding Abstract | Although genetic association studies have been with us for many years, even for the simplest analyses there is little consensus on the most appropriate statistical procedures. Here I give an overview of statistical approaches to population association studies, including preliminary analyses (Hardy–Weinberg equilibrium testing, inference of phase and missing data, and SNP tagging), and single-SNP and multipoint tests for association. My goal is to outline the key methods with a brief discussion of problems (population structure and multiple testing), avenues for solutions and some ongoing developments. Haplotype The goal of population association studies is to identify will obtain a clearer picture of the statistical issues and A combination of alleles at patterns of polymorphisms that vary systematically gain some ideas for new or modified approaches. different loci on the same between individuals with different disease states and In this review I cover only population association chromosome. could therefore represent the effects of risk-enhancing or studies in which unrelated individuals of different dis- protective alleles (BOXES 1,2). That sounds easy enough: ease states are typed at a number of SNP markers. I do Population stratification Refers to a situation in which what could be difficult about spotting allele patterns that not address family-based association studies, admixture the population of interest are overrepresented in cases relative to controls? mapping or linkage studies (BOX 3), which also have an includes subgroups of One fundamental problem is that the genome is so important role in efforts to understand the effects of individuals that are on average large that patterns that are suggestive of a causal poly- genes on disease2.