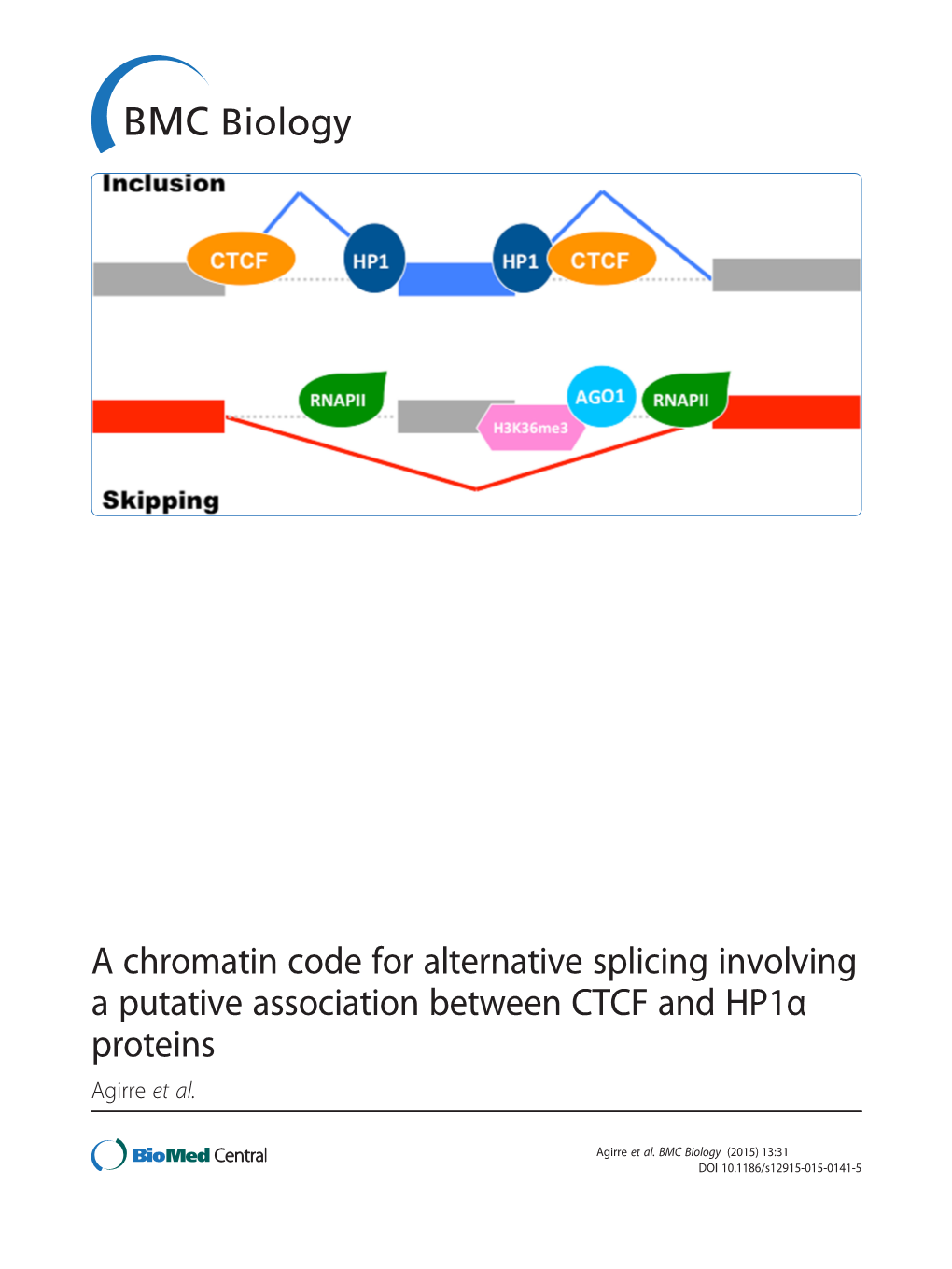
A Chromatin Code for Alternative Splicing Involving a Putative Association Between CTCF and Hp1α Proteins Agirre Et Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Alternative Role of DNA Methylation in Splicing Regulation
TIGS-1191; No. of Pages 7 Review The alternative role of DNA methylation in splicing regulation Galit Lev Maor, Ahuvi Yearim, and Gil Ast Department of Human Molecular Genetics and Biochemistry, Sackler Medical School, Tel Aviv University, Tel Aviv, Israel Although DNA methylation was originally thought to only Alternative splicing is an evolutionarily conserved mech- affect transcription, emerging evidence shows that it also anism that increases transcriptome and proteome diversity regulates alternative splicing. Exons, and especially splice by allowing the generation of multiple mRNA products from sites, have higher levels of DNA methylation than flanking a single gene [10]. More than 90% of human genes were introns, and the splicing of about 22% of alternative exons shown to undergo alternative splicing [11,12]. Furthermore, is regulated by DNA methylation. Two different mecha- the average number of spliced isoforms per gene is higher in nisms convey DNA methylation information into the vertebrates [13], implying that the prevalence of alternative regulation of alternative splicing. The first involves mod- splicing in these organisms is important for their greater ulation of the elongation rate of RNA polymerase II (Pol II) complexity. The splicing reaction is regulated by various by CCCTC-binding factor (CTCF) and methyl-CpG binding activating and repressing elements such as cis-acting se- protein 2 (MeCP2); the second involves the formation of a quence signals and RNA-binding proteins [13–15]. Its regu- protein bridge by heterochromatin protein 1 (HP1) that lation is essential for providing cells and tissues their recruits splicing factors onto transcribed alternative specific features, and for their response to environmental exons. -

A Newly Assigned Role of CTCF in Cellular Response to Broken Dnas
biomolecules Review A Newly Assigned Role of CTCF in Cellular Response to Broken DNAs Mi Ae Kang and Jong-Soo Lee * Department of Life Sciences, Ajou University, Suwon 16499, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-31-219-1886; Fax: +82-31-219-1615 Abstract: Best known as a transcriptional factor, CCCTC-binding factor (CTCF) is a highly con- served multifunctional DNA-binding protein with 11 zinc fingers. It functions in diverse genomic processes, including transcriptional activation/repression, insulation, genome imprinting and three- dimensional genome organization. A big surprise has recently emerged with the identification of CTCF engaging in the repair of DNA double-strand breaks (DSBs) and in the maintenance of genome fidelity. This discovery now adds a new dimension to the multifaceted attributes of this protein. CTCF facilitates the most accurate DSB repair via homologous recombination (HR) that occurs through an elaborate pathway, which entails a chain of timely assembly/disassembly of various HR-repair complexes and chromatin modifications and coordinates multistep HR processes to faithfully restore the original DNA sequences of broken DNA sites. Understanding the functional crosstalks between CTCF and other HR factors will illuminate the molecular basis of various human diseases that range from developmental disorders to cancer and arise from impaired repair. Such knowledge will also help understand the molecular mechanisms underlying the diverse functions of CTCF in genome biology. In this review, we discuss the recent advances regarding this newly assigned versatile role of CTCF and the mechanism whereby CTCF functions in DSB repair. Keywords: CTCF; DNA damage repair; homologous recombination Citation: Kang, M.A.; Lee, J.-S. -

CCCTC-Binding Factor Is Essential to the Maintenance and Quiescence of Hematopoietic Stem Cells in Mice
OPEN Experimental & Molecular Medicine (2017) 49, e371; doi:10.1038/emm.2017.124 Official journal of the Korean Society for Biochemistry and Molecular Biology www.nature.com/emm ORIGINAL ARTICLE CCCTC-binding factor is essential to the maintenance and quiescence of hematopoietic stem cells in mice Tae-Gyun Kim1,2,3,4, Sueun Kim1,2,4, Soyeon Jung1, Mikyoung Kim1, Bobae Yang1,2, Min-Geol Lee2,3 and Hyoung-Pyo Kim1,2 Hematopoiesis involves a series of lineage differentiation programs initiated in hematopoietic stem cells (HSCs) found in bone marrow (BM). To ensure lifelong hematopoiesis, various molecular mechanisms are needed to maintain the HSC pool. CCCTC-binding factor (CTCF) is a DNA-binding, zinc-finger protein that regulates the expression of its target gene by organizing higher order chromatin structures. Currently, the role of CTCF in controlling HSC homeostasis is unknown. Using a tamoxifen- inducible CTCF conditional knockout mouse system, we aimed to determine whether CTCF regulates the homeostatic maintenance of HSCs. In adult mice, acute systemic CTCF ablation led to severe BM failure and the rapid shrinkage of multiple c-Kithi progenitor populations, including Sca-1+ HSCs. Similarly, hematopoietic system-confined CTCF depletion caused an acute loss of HSCs and highly increased mortality. Mixed BM chimeras reconstituted with supporting BM demonstrated that CTCF deficiency-mediated HSC depletion has both cell-extrinsic and cell-intrinsic effects. Although c-Kithi myeloid progenitor cell populations were severely reduced after ablating Ctcf, c-Kitint common lymphoid progenitors and their progenies were less affected by the lack of CTCF. Whole-transcriptome microarray and cell cycle analyses indicated that CTCF deficiency results in the enhanced expression of the cell cycle-promoting program, and that CTCF-depleted HSCs express higher levels of reactive oxygen species (ROS). -

Enhancement of Transgene Expression by NF-Y and CTCF Devon Zimmerman , Krupa Patel , Matthew Hall , & Jacob Elmer
bioRxiv preprint doi: https://doi.org/10.1101/336156; this version posted May 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Enhancement of Transgene Expression by NF-Y and CTCF Devon Zimmermana, Krupa Patela, Matthew Halla, & Jacob Elmera a Department of Chemical Engineering Villanova University 800 East Lancaster Avenue Villanova, PA, USA 19085 *Corresponding Author: Dr. Jacob Elmer 119 White Hall Department of Chemical Engineering 800 East Lancaster Avenue Villanova, PA 19085 [email protected] (610) 519-3093 (P) (610) 519-5859 (F) bioRxiv preprint doi: https://doi.org/10.1101/336156; this version posted May 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract If a transgene is effectively delivered to a cell, its expression may still be limited by epigenetic mechanisms that silence the transgene. Indeed, once the transgene reaches the nucleus, it may be bound by histone proteins and condensed into heterochromatin or associated with repressor proteins that block transcription. In this study, we sought to enhance transgene expression by adding binding motifs for several different epigenetic enzymes either upstream or downstream of two promoters (CMV and EF1α). Screening these plasmids revealed that luciferase expression was enhanced 10-fold by the addition of a CCAAT box just upstream of the EF1α promoter to recruit nuclear transcription factor Y (NF-Y), while inserting a CCCTC- binding factor (CTCF) motif downstream of the EF1α promoter enhanced expression 14-fold (14.03 ± 6.54). -

The Role of CTCF in Cell Differentiation Rodrigo G
© 2018. Published by The Company of Biologists Ltd | Development (2018) 145, dev137729. doi:10.1242/dev.137729 REVIEW Developing in 3D: the role of CTCF in cell differentiation Rodrigo G. Arzate-Mejıá1,Félix Recillas-Targa1 and Victor G. Corces2,* ABSTRACT precise role played by CTCF during organismal development has CTCF is a highly conserved zinc-finger DNA-binding protein that remained poorly explored. Here, we review evidence linking CTCF mediates interactions between distant sequences in the genome. As to the control of developmental processes (summarized in Table 1). a consequence, CTCF regulates enhancer-promoter interactions and We first provide an overview of how CTCF functions to regulate contributes to the three-dimensional organization of the genome. genome 3D organization and how this role affects gene expression. Recent studies indicate that CTCF is developmentally regulated, We then discuss the roles of CTCF in the development and suggesting that it plays a role in cell type-specific genome differentiation of various cell and tissue types, ranging from organization. Here, we review these studies and discuss how CTCF embryonic stem cells (ESCs) to neural, cardiac and muscle cells. We functions during the development of various cell and tissue types, conclude with an integrative view of CTCF as an important ranging from embryonic stem cells and gametes, to neural, muscle determinant of cell lineage specification during vertebrate and cardiac cells. We propose that the lineage-specific control of development. CTCF levels, and its partnership with lineage-specific transcription factors, allows for the control of cell type-specific gene expression via Mechanisms of CTCF function chromatin looping. -

Th2 Cytokine Expression CCCTC-Binding Factor in The
Critical Role for the Transcription Regulator CCCTC-Binding Factor in the Control of Th2 Cytokine Expression This information is current as Claudia Ribeiro de Almeida, Helen Heath, Sanja Krpic, of October 1, 2021. Gemma M. Dingjan, Jan Piet van Hamburg, Ingrid Bergen, Suzanne van de Nobelen, Frank Sleutels, Frank Grosveld, Niels Galjart and Rudi W. Hendriks J Immunol 2009; 182:999-1010; ; doi: 10.4049/jimmunol.182.2.999 http://www.jimmunol.org/content/182/2/999 Downloaded from References This article cites 57 articles, 14 of which you can access for free at: http://www.jimmunol.org/content/182/2/999.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on October 1, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Critical Role for the Transcription Regulator CCCTC-Binding Factor in the Control of Th2 Cytokine Expression1 Claudia Ribeiro de Almeida,2*† Helen Heath,2‡ Sanja Krpic,‡ Gemma M. -

Exploration of CTCF Post-Translation Modifications Uncovers Serine-224 Phosphorylation by PLK1 at Pericentric Regions During
TOOLS AND RESOURCES Exploration of CTCF post-translation modifications uncovers Serine-224 phosphorylation by PLK1 at pericentric regions during the G2/M transition Brian C Del Rosario1,2†, Andrea J Kriz1,2†, Amanda M Del Rosario3, Anthony Anselmo4, Christopher J Fry5, Forest M White3, Ruslan I Sadreyev4, Jeannie T Lee1,2* 1Department of Molecular Biology, Howard Hughes Medical Institute, Massachusetts General Hospital, Boston, United States; 2Department of Genetics, Harvard Medical School, Boston, United States; 3Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, United States; 4Department of Molecular Biology, Massachusetts General Hospital, Boston, United States; 5Cell Signaling Technology, Danvers, United States Abstract The zinc finger CCCTC-binding protein (CTCF) carries out many functions in the cell. Although previous studies sought to explain CTCF multivalency based on sequence composition of binding sites, few examined how CTCF post-translational modification (PTM) could contribute to function. Here, we performed CTCF mass spectrometry, identified a novel phosphorylation site at Serine 224 (Ser224-P), and demonstrate that phosphorylation is carried out by Polo-like kinase 1 (PLK1). CTCF Ser224-P is chromatin-associated, mapping to at least a subset of known CTCF sites. CTCF Ser224-P accumulates during the G2/M transition of the cell cycle and is enriched at pericentric regions. The phospho-obviation mutant, S224A, appeared normal. However, the *For correspondence: phospho-mimic mutant, S224E, is detrimental to mouse embryonic stem cell colonies. While ploidy [email protected] and chromatin architecture appear unaffected, S224E mutants differentially express hundreds of †These authors contributed genes, including p53 and p21. We have thus identified a new CTCF PTM and provided evidence of equally to this work biological function. -

Discovery of Biased Orientation of Human DNA Motif Sequences
bioRxiv preprint doi: https://doi.org/10.1101/290825; this version posted January 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Discovery of biased orientation of human DNA motif sequences 2 affecting enhancer-promoter interactions and transcription of genes 3 4 Naoki Osato1* 5 6 1Department of Bioinformatic Engineering, Graduate School of Information Science 7 and Technology, Osaka University, Osaka 565-0871, Japan 8 *Corresponding author 9 E-mail address: [email protected], [email protected] 10 1 bioRxiv preprint doi: https://doi.org/10.1101/290825; this version posted January 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 11 Abstract 12 Chromatin interactions have important roles for enhancer-promoter interactions 13 (EPI) and regulating the transcription of genes. CTCF and cohesin proteins are located 14 at the anchors of chromatin interactions, forming their loop structures. CTCF has 15 insulator function limiting the activity of enhancers into the loops. DNA binding 16 sequences of CTCF indicate their orientation bias at chromatin interaction anchors – 17 forward-reverse (FR) orientation is frequently observed. DNA binding sequences of 18 CTCF were found in open chromatin regions at about 40% - 80% of chromatin 19 interaction anchors in Hi-C and in situ Hi-C experimental data. -

The Vitamin D Receptor: Contemporary Genomic Approaches Reveal New Basic and Translational Insights
REVIEW SERIES: NUCLEAR RECEPTORS The Journal of Clinical Investigation Series Editor: Mitchell A. Lazar The vitamin D receptor: contemporary genomic approaches reveal new basic and translational insights J. Wesley Pike, Mark B. Meyer, Seong-Min Lee, Melda Onal, and Nancy A. Benkusky Department of Biochemistry, University of Wisconsin — Madison, Madison, Wisconsin, USA. The vitamin D receptor (VDR) is the single known regulatory mediator of hormonal 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] in higher vertebrates. It acts in the nucleus of vitamin D target cells to regulate the expression of genes whose products control diverse, cell type–specific biological functions that include mineral homeostasis. In this Review we describe progress that has been made in defining new cellular sites of action of this receptor, the mechanisms through which this mediator controls the expression of genes, the biology that ensues, and the translational impact of this receptor on human health and disease. We conclude with a brief discussion of what comes next in understanding vitamin D biology and the mechanisms that underlie its actions. Introduction involved (14, 15). With this background, we then comment briefly on In the early 1970s, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] was the current translational impact of several features of VDR action identified as both the exclusive, metabolically active form of vita- and function on human health and disease. min D and a key component of what proved to be an exquisite endo- crine system that regulates numerous biologic processes in higher The VDR vertebrates (1, 2). This pioneering discovery ended a decades-long VDR tissue distribution. -

Proceedings of the 5Th International Symposium on Vitamin D and Analogs in Cancer Prevention and Therapy
ANTICANCER RESEARCH 35: 1143-1218 (2015) PROCEEDINGS OF THE 5TH INTERNATIONAL SYMPOSIUM ON VITAMIN D AND ANALOGS IN CANCER PREVENTION AND THERAPY 2-3 May, 2014 Krefeld, Germany Local Organizing Committee J. Reichrath1 M. Friedrich2 Th. Vogt1 1Department of Dermatology, Venerology and Allergology, The Saarland University Hospital, Homburg/Saar, Germany; 2Department of Obstetrics and Gynecology, HELIOS Hospital, Krefeld, Germany ANTICANCER RESEARCH 35: 1143-1152 (2015) Review What Do We Learn from the Genome-wide Perspective on Vitamin D3? CARSTEN CARLBERG School of Medicine, Institute of Biomedicine, University of Eastern Finland, Kuopio, Finland Abstract. Vitamin D3 insufficiency is associated with a cell type that sum-up to more than 23,000 non-overlapping number of diseases, such as cancer and autoimmune loci of the receptor. After ligand stimulation VDR associates disorders. This important medical problem leads to the with many new binding loci, of which the most important have question, whether an insight into the genome-wide actions of a higher rate of DR3-type VDR binding sequences than the transcription factor vitamin D receptor (VDR) and its high average sites. On the majority of latter VDR interacts directly affinity ligand 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3) can or indirectly with genomic DNA in a presently uncharacterized help in a more global appreciation of the physiological impact fashion. Formaldehyde-assisted isolation of regulatory of vitamin D3. Chromatin immunoprecipitation sequencing elements sequencing (FAIRE-seq) monitors the dynamically (ChIP-seq) studies in 6 human cell culture models opening chromatin regions after 1,25(OH)2D3 stimulation. The demonstrated 1,000 to 10,000 genomic VDR binding sites per integration of ChIP-seq and FAIRE-seq data combined with a screening for DR3-type sequences facilitates the identification of key VDR binding sites and primary 1,25(OH)2D3 target genes. -

Endothelial Cell–Glucocorticoid Receptor Interactions and Regulation of Wnt Signaling
RESEARCH ARTICLE Endothelial cell–glucocorticoid receptor interactions and regulation of Wnt signaling Han Zhou,1,2 Sameet Mehta,3 Swayam Prakash Srivastava,1,2 Kariona Grabinska,2,4 Xinbo Zhang,2,5,6 Chris Wong,1 Ahmad Hedayat,1,2 Paola Perrotta,2,4 Carlos Fernández-Hernando,2,5,6,7 William C. Sessa,2,4 and Julie E. Goodwin1,2 1Department of Pediatrics, 2Vascular Biology and Therapeutics Program, 3Department of Genetics, 4Department of Pharmacology, 5Integrative Cell Signaling and Neurobiology of Metabolism Program, 6Department of Comparative Medicine, and 7Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA. Vascular inflammation is present in many cardiovascular diseases, and exogenous glucocorticoids have traditionally been used as a therapy to suppress inflammation. However, recent data have shown that endogenous glucocorticoids, acting through the endothelial glucocorticoid receptor, act as negative regulators of inflammation. Here, we performed ChIP for the glucocorticoid receptor, followed by next-generation sequencing in mouse endothelial cells to investigate how the endothelial glucocorticoid receptor regulates vascular inflammation. We identified a role of the Wnt signaling pathway in this setting and show that loss of the endothelial glucocorticoid receptor results in upregulation of Wnt signaling both in vitro and in vivo using our validated mouse model. Furthermore, we demonstrate glucocorticoid receptor regulation of a key gene in the Wnt pathway, Frzb, via a glucocorticoid response element gleaned from our genomic data. These results suggest a role for endothelial Wnt signaling modulation in states of vascular inflammation. Introduction Inflammation is a complex cascade of adaptive cellular responses to injurious stimuli, which occurs in many cardiovascular diseases (1). -

Nucleosome Dynamics of Human Ipsc During the Early Stages of Neurodevelopment
bioRxiv preprint doi: https://doi.org/10.1101/398792; this version posted August 23, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Nucleosome dynamics of human iPSC during the early stages of neurodevelopment Janet C Harwood1, Nicholas A Kent2, Nicholas D Allen2 and Adrian J Harwood2,3* 1 MRC Centre for Neuropsychiatric Genetics & Genomics, Cardiff University, Maindy Rd, Cardiff CF24 4HQ, UK 2 School of Biosciences, Cardiff University, Museum Ave, Cardiff CF10 3AX, UK 3 Neuroscience and Mental Health Research Institute (NMHRI), Cardiff University, Maindy Rd, Cardiff CF24 4HQ, UK • Correspondence: [email protected] Key Words: Nucleosome Positioning, Chromatin, human iPSC, neurodevelopment. 1 bioRxiv preprint doi: https://doi.org/10.1101/398792; this version posted August 23, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Regulation of nucleosome positioning is important for neurodevelopment, and mutation of genes mediating chromatin remodelling are strongly associated with human neurodevelopmental disorders. Unicellular organisms possess arrays of highly positioned nucleosomes within their chromatin, occupying up to 80% of their genomes. These span gene-coding and regulatory regions, and can be associated with local changes of gene transcription. In the much larger genome of human cells, the roles of nucleosome positioning are less clear, and this raises questions of how nucleosome dynamics interfaces with human neurodevelopment.