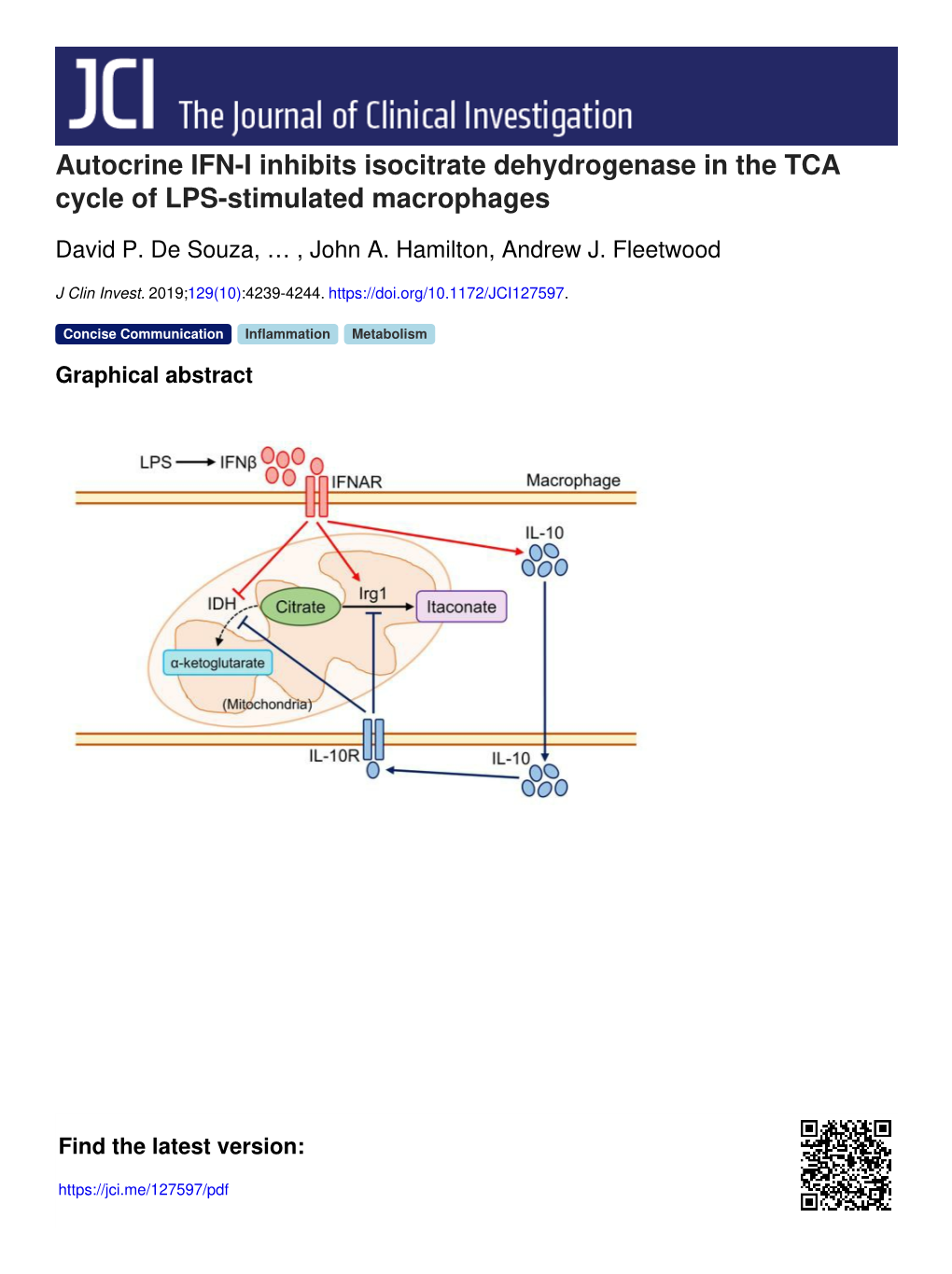
Autocrine IFN-I Inhibits Isocitrate Dehydrogenase in the TCA Cycle of LPS-Stimulated Macrophages
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download (Pdf)
Invivoscribe's wholly-owned Laboratories for Personalized Molecular LabPMM LLC Medicine® (LabPMM) is a network of international reference laboratories that provide the medical and pharmaceutical communities with worldwide Located in San Diego, California, USA, it holds access to harmonized and standardized clinical testing services. We view the following accreditations and certifications: internationally reproducible and concordant testing as a requirement for ISO 15189, CAP, and CLIA, and is licensed to provide diagnostic consistent stratification of patients for enrollment in clinical trials, and the laboratory services in the states of California, Florida, foundation for establishing optimized treatment schedules linked to patient’s Maryland, New York, Pennsylvania, and Rhode Island. individual profile. LabPMM provides reliable patient stratification at diagnosis LabPMM GmbH and monitoring, throughout the entire course of treatment in support of Personalized Molecular Medicine® and Personalized Based in Martinsried (Munich), Germany. It is an ISO 15189 Molecular Diagnostics®. accredited international reference laboratory. CLIA/CAP accreditation is planned. Invivoscribe currently operates four clinical laboratories to serve partners in the USA (San Diego, CA), Europe (Munich, Germany), and Asia (Tokyo, Japan and Shanghai, China). These laboratories use the same critical LabPMM 合同会社 reagents and software which are developed consistently with ISO Located in Kawasaki (Tokyo), Japan and a licensed clinical lab. 13485 design control. Our cGMP reagents, rigorous standards for assay development & validation, and testing performed consistently under ISO 15189 requirements help ensure LabPMM generates standardized and concordant test results worldwide. Invivoscribe Diagnostic Technologies (Shanghai) Co., Ltd. LabPMM is an international network of PersonalMed Laboratories® focused on molecular oncology biomarker studies. Located in Shangai, China. -

IDH2 Mutations—Co-Opting Cellular Metabolism for Malignant Transformation Eytan M
Published OnlineFirst November 9, 2015; DOI: 10.1158/1078-0432.CCR-15-0362 Molecular Pathways Clinical Cancer Research Molecular Pathways: IDH2 Mutations—Co-opting Cellular Metabolism for Malignant Transformation Eytan M. Stein Abstract Mutations in mitochondrial IDH2, one of the three isoforms function that catalyzes the conversion of alpha-ketoglutarate to of IDH, were discovered in patients with gliomas in 2009 and beta-hydroxyglutarate (2-HG). Supranormal levels of 2-HG subsequently described in acute myelogenous leukemia (AML), lead to hypermethylation of epigenetic targets and a subse- angioimmunoblastic T-cell lymphoma, chondrosarcoma, and quent block in cellular differentiation. AG-221, a small-mole- intrahepatic chloangiocarcinoma. The effects of mutations in cule inhibitor of mutant IDH2, is being explored in a phase I IDH2 on cellular metabolism, the epigenetic state of mutated clinical trial for the treatment of AML, other myeloid malig- cells, and cellular differentiation have been elucidated in vitro nancies, solid tumors, and gliomas. Clin Cancer Res; 22(1); 16–19. and in vivo.MutationsinIDH2 lead to an enzymatic gain of Ó2015 AACR. Background Interestingly, in a screen of 398 patients with AML, TET2 and IDH2 mutations were mutually exclusive, suggesting that these Isocitrate dehydrogenase (IDH) catalyzes the conversion of two mutations have a similar function. Wild-type TET2 demethy- isocitrate to alpha-ketoglutarate. IDH occurs in three isoforms, lates DNA, and mutations in TET2 lead to increases in 5-methyl- IDH1, located in the cytoplasm, IDH2 located in the mitochon- cytosine similar to those seen in patients with IDH mutations. It dria, and IDH3, which functions as part of the tricarboxylic acid has been suggested that elevated levels of 2-HG caused by mutant cycle. -

Effect of Citric Acid Cycle Genetic Variants and Their Interactions With
cancers Article Effect of Citric Acid Cycle Genetic Variants and Their Interactions with Obesity, Physical Activity and Energy Intake on the Risk of Colorectal Cancer: Results from a Nested Case-Control Study in the UK Biobank Sooyoung Cho 1 , Nan Song 2,3 , Ji-Yeob Choi 2,4,5 and Aesun Shin 1,2,* 1 Department of Preventive Medicine, Seoul National University College of Medicine, Seoul 03080, Korea; [email protected] 2 Cancer Research Institute, Seoul National University, Seoul 03080, Korea; [email protected] (N.S.); [email protected] (J.-Y.C.) 3 Department of Epidemiology and Cancer Control, St. Jude Children’s Research Hospital, Memphis, TN 38105, USA 4 Department of Biomedical Sciences, Graduate School of Seoul National University, Seoul 03080, Korea 5 Medical Research Center, Institute of Health Policy and Management, Seoul National University, Seoul 03080, Korea * Correspondence: [email protected]; Tel.: +82-2-740-8331; Fax: +82-2-747-4830 Received: 18 August 2020; Accepted: 9 October 2020; Published: 12 October 2020 Simple Summary: The citric acid cycle has a central role in the cellular energy metabolism and biosynthesis of macromolecules in the mitochondrial matrix. We identified the single nucleotide polymorphisms (SNPs) of the citrate acid cycle with colorectal cancer susceptibility in UK population. Furthermore, we found the significant interaction of SNPs in the citric acid cycle with the contributors to energy balance and SNP-SNP interactions. Our findings provide clues to the etiology in cancer development related to energy metabolism and evidence on identification of the population at high risk of colorectal cancer. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Mtor Coordinates Transcriptional Programs and Mitochondrial Metabolism of Activated Treg Subsets to Protect Tissue Homeostasis
ARTICLE DOI: 10.1038/s41467-018-04392-5 OPEN mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis Nicole M. Chapman1, Hu Zeng 1, Thanh-Long M. Nguyen1, Yanyan Wang1, Peter Vogel 2, Yogesh Dhungana1, Xiaojing Liu3, Geoffrey Neale 4, Jason W. Locasale 3 & Hongbo Chi1 1234567890():,; Regulatory T (Treg) cells derived from the thymus (tTreg) and periphery (pTreg) have central and distinct functions in immunosuppression, but mechanisms for the generation and acti- vation of Treg subsets in vivo are unclear. Here, we show that mechanistic target of rapamycin (mTOR) unexpectedly supports the homeostasis and functional activation of tTreg and pTreg cells. mTOR signaling is crucial for programming activated Treg-cell function to protect immune tolerance and tissue homeostasis. Treg-specific deletion of mTOR drives sponta- neous effector T-cell activation and inflammation in barrier tissues and is associated with reduction in both thymic-derived effector Treg (eTreg) and pTreg cells. Mechanistically, mTOR functions downstream of antigenic signals to drive IRF4 expression and mitochondrial metabolism, and accordingly, deletion of mitochondrial transcription factor A (Tfam) severely impairs Treg-cell suppressive function and eTreg-cell generation. Collectively, our results show that mTOR coordinates transcriptional and metabolic programs in activated Treg subsets to mediate tissue homeostasis. 1 Department of Immunology, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, MS 351, Memphis, TN 38105, USA. 2 Department of Pathology, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, MS 250, Memphis, TN 38105, USA. 3 Department of Pharmacology & Cancer Biology, Duke University School of Medicine, Levine Science Research Center C266, Box 3813, Durham, NC 27710, USA. -

Anti-IDH3A Antibody (ARG42205)
Product datasheet [email protected] ARG42205 Package: 100 μl anti-IDH3A antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes IDH3A Tested Reactivity Hu, Ms, Rat Tested Application ICC/IF, IHC-P, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name IDH3A Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 28-366 of Human IDH3A (NP_005521.1). Conjugation Un-conjugated Alternate Names Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial; EC 1.1.1.41; NAD; Isocitric dehydrogenase subunit alpha; + Application Instructions Application table Application Dilution ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control Raji Calculated Mw 40 kDa Observed Size ~ 40 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw www.arigobio.com 1/3 cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol IDH3A Gene Full Name isocitrate dehydrogenase 3 (NAD+) alpha Background Isocitrate dehydrogenases catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate. -

Src-Family Kinases Impact Prognosis and Targeted Therapy in Flt3-ITD+ Acute Myeloid Leukemia
Src-Family Kinases Impact Prognosis and Targeted Therapy in Flt3-ITD+ Acute Myeloid Leukemia Title Page by Ravi K. Patel Bachelor of Science, University of Minnesota, 2013 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2019 Commi ttee Membership Pa UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE Commi ttee Membership Page This dissertation was presented by Ravi K. Patel It was defended on May 31, 2019 and approved by Qiming (Jane) Wang, Associate Professor Pharmacology and Chemical Biology Vaughn S. Cooper, Professor of Microbiology and Molecular Genetics Adrian Lee, Professor of Pharmacology and Chemical Biology Laura Stabile, Research Associate Professor of Pharmacology and Chemical Biology Thomas E. Smithgall, Dissertation Director, Professor and Chair of Microbiology and Molecular Genetics ii Copyright © by Ravi K. Patel 2019 iii Abstract Src-Family Kinases Play an Important Role in Flt3-ITD Acute Myeloid Leukemia Prognosis and Drug Efficacy Ravi K. Patel, PhD University of Pittsburgh, 2019 Abstract Acute myelogenous leukemia (AML) is a disease characterized by undifferentiated bone-marrow progenitor cells dominating the bone marrow. Currently the five-year survival rate for AML patients is 27.4 percent. Meanwhile the standard of care for most AML patients has not changed for nearly 50 years. We now know that AML is a genetically heterogeneous disease and therefore it is unlikely that all AML patients will respond to therapy the same way. Upregulation of protein-tyrosine kinase signaling pathways is one common feature of some AML tumors, offering opportunities for targeted therapy. -

Kras, Braf, Erbb2, Tp53
Supplementary Table 1. SBT-EOC cases used for molecular analyses Fresh Frozen Tissue FFPE Tissue Tumour HRM Case ID PCR-Seq HRM component Oncomap (KRAS, BRAF, CNV GE CNV IHC (NRAS) (TP53 Only) ERBB2, TP53) Paired Cases 15043 SBT & INV √ √ √ √ √ √ 65662 SBT & INV √ √ √ √ √ 65661 SBT & INV √ √ √ √ √ √ 9128 SBT & INV √ √ √ √ √ 2044 SBT & INV √ √ √ √ √ 3960 SBT & INV √ √ √ √ √ 5899 SBT & INV √ √ √ √ √ 65663 SBT & INV √ √ √ Inv √ √ √ 65666 SBT √ Inv √ √ √ √ √ 65664 SBT √ √ √ Inv √ √ √ √ √ 15060 SBT √ √ √ Inv √ √ √ √ √ 65665 SBT √ √ √ Inv √ √ √ √ √ 65668 SBT √ √ √ Inv √ √ √ √ 65667 SBT √ √ √ Inv √ √ √ √ 15018 SBT √ Inv √ √ √ √ 15071 SBT √ Inv √ √ √ √ 65670 SBT √ Inv √ √ √ 8982 SBT √ Unpaired Cases 15046 Inv √ √ √ 15014 Inv √ √ √ 65671 Inv √ √ √ 65672 SBT √ √ √ 65673 Inv √ √ √ 65674 Inv √ √ √ 65675 Inv √ √ √ 65676 Inv √ √ √ 65677 Inv √ √ √ 65678 Inv √ √ √ 65679 Inv √ √ √ Fresh Frozen Tissue FFPE Tissue Tumour HRM Case ID PCR-Seq HRM component Oncomap (KRAS, BRAF, CNV GE CNV IHC (NRAS) (TP53 Only) ERBB2, TP53) 65680 Inv √ √ √ 5349 Inv √ √ √ 7957 Inv √ √ √ 8395 Inv √ √ √ 8390 Inv √ √ 2110 Inv √ √ 6328 Inv √ √ 9125 Inv √ √ 9221 Inv √ √ 10701 Inv √ √ 1072 Inv √ √ 11266 Inv √ √ 11368 Inv √ √ √ 12237 Inv √ √ 2064 Inv √ √ 3539 Inv √ √ 2189 Inv √ √ √ 5711 Inv √ √ 6251 Inv √ √ 6582 Inv √ √ 7200 SBT √ √ √ 8633 Inv √ √ √ 9579 Inv √ √ 10740 Inv √ √ 11766 Inv √ √ 3958 Inv √ √ 4723 Inv √ √ 6244 Inv √ √ √ 7716 Inv √ √ SBT-EOC, serous carcinoma with adjacent borderline regions; SBT, serous borderline component of SBT- EOC; Inv, invasive component of SBT-EOC; -

Reprogramming of Isocitrate Dehydrogenases Expression and Activity by the Androgen
Author Manuscript Published OnlineFirst on May 8, 2019; DOI: 10.1158/1541-7786.MCR-19-0020 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. 1 Reprogramming of isocitrate dehydrogenases expression and activity by the androgen 2 receptor in prostate cancer 3 4 Kevin Gonthier1,2, Raghavendra Tejo Karthik Poluri1,2, Cindy Weidmann1, Maude Tadros1, and 5 Étienne Audet-Walsh1,2,3 6 7 1 Endocrinology - Nephrology Research Axis, Centre de recherche du CHU de Québec - 8 Université Laval, Québec City, Canada 9 2 Department of molecular medicine, Faculty of Medicine, Université Laval, Québec City, 10 Canada 11 3 Centre de recherche sur le cancer de l’Université Laval, Québec City, Canada 12 13 Keywords: steroid; metabolism; glioma; nuclear receptor; castration-resistant prostate cancer, 14 IDH1, oncometabolism 15 16 Abbreviated title: Reprogramming of IDH activity by AR in PCa 17 18 Corresponding Author: Étienne Audet-Walsh, Centre de recherche du CHU de Québec, 2705 19 Boulevard Laurier, room R-4714, Québec City, QC, Canada, G1V 4G2 20 Phone: 1-418-525-4444 ext. 48678 ; e-mail : [email protected] 21 22 Disclosure statement: the authors declare no potential conflicts of interest. 1 Downloaded from mcr.aacrjournals.org on September 24, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on May 8, 2019; DOI: 10.1158/1541-7786.MCR-19-0020 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. 23 Abstract 24 Mutations of the isocitrate dehydrogenase genes IDH1 and IDH2, key enzymes involved in 25 citrate metabolism, are important oncogenic events in several cancer types, including in 1-3% of 26 all prostate cancer (PCa) cases. -

Potential Genotoxicity from Integration Sites in CLAD Dogs Treated Successfully with Gammaretroviral Vector-Mediated Gene Therapy
Gene Therapy (2008) 15, 1067–1071 & 2008 Nature Publishing Group All rights reserved 0969-7128/08 $30.00 www.nature.com/gt SHORT COMMUNICATION Potential genotoxicity from integration sites in CLAD dogs treated successfully with gammaretroviral vector-mediated gene therapy M Hai1,3, RL Adler1,3, TR Bauer Jr1,3, LM Tuschong1, Y-C Gu1,XWu2 and DD Hickstein1 1Experimental Transplantation and Immunology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, USA and 2Laboratory of Molecular Technology, Scientific Applications International Corporation-Frederick, National Cancer Institute-Frederick, Frederick, Maryland, USA Integration site analysis was performed on six dogs with in hematopoietic stem cells. Integrations clustered around canine leukocyte adhesion deficiency (CLAD) that survived common insertion sites more frequently than random. greater than 1 year after infusion of autologous CD34+ bone Despite potential genotoxicity from RIS, to date there has marrow cells transduced with a gammaretroviral vector been no progression to oligoclonal hematopoiesis and no expressing canine CD18. A total of 387 retroviral insertion evidence that vector integration sites influenced cell survival sites (RIS) were identified in the peripheral blood leukocytes or proliferation. Continued follow-up in disease-specific from the six dogs at 1 year postinfusion. A total of 129 RIS animal models such as CLAD will be required to provide an were identified in CD3+ T-lymphocytes and 102 RIS in accurate estimate -

As a Potential Therapeutic Approach for AML Wittawat Chantkran 1,2,Ya-Chinghsieh1, Daniella Zheleva3,Sheelaghframe3, Helen Wheadon1 and Mhairi Copland 1
Chantkran et al. Cell Death Discovery (2021) 7:137 https://doi.org/10.1038/s41420-021-00496-y Cell Death Discovery ARTICLE Open Access Interrogation of novel CDK2/9 inhibitor fadraciclib (CYC065) as a potential therapeutic approach for AML Wittawat Chantkran 1,2,Ya-ChingHsieh1, Daniella Zheleva3,SheelaghFrame3, Helen Wheadon1 and Mhairi Copland 1 Abstract Over the last 50 years, there has been a steady improvement in the treatment outcome of acute myeloid leukemia (AML). However, median survival in the elderly is still poor due to intolerance to intensive chemotherapy and higher numbers of patients with adverse cytogenetics. Fadraciclib (CYC065), a novel cyclin-dependent kinase (CDK) 2/9 inhibitor, has preclinical efficacy in AML. In AML cell lines, myeloid cell leukemia 1 (MCL-1) was downregulated following treatment with fadraciclib, resulting in a rapid induction of apoptosis. In addition, RNA polymerase II (RNAPII)-driven transcription was suppressed, rendering a global gene suppression. Rapid induction of apoptosis was observed in primary AML cells after treatment with fadraciclib for 6–8 h. Twenty-four hours continuous treatment further increased efficacy of fadraciclib. Although preliminary results showed that AML cell lines harboring KMT2A rearrangement (KMT2A-r) are more sensitive to fadraciclib, we found that the drug can induce apoptosis and decrease MCL-1 expression in primary AML cells, regardless of KMT2A status. Importantly, the diversity of genetic mutations observed in primary AML patient samples was associated with variable response to fadraciclib, confirming the need for fi 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; patient strati cation to enable a more effective and personalized treatment approach. Synergistic activity was demonstrated when fadraciclib was combined with the BCL-2 inhibitor venetoclax, or the conventional chemotherapy agents, cytarabine, or azacitidine, with the combination of fadraciclib and azacitidine having the most favorable therapeutic window. -

Mouse Idh3a Mutations Cause Retinal Degeneration and Reduced Mitochondrial Function Amy S
© 2018. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2018) 11, dmm036426. doi:10.1242/dmm.036426 RESEARCH ARTICLE Mouse Idh3a mutations cause retinal degeneration and reduced mitochondrial function Amy S. Findlay1, Roderick N. Carter2, Becky Starbuck3, Lisa McKie1, Klára Nováková1, Peter S. Budd1, Margaret A. Keighren1, Joseph A. Marsh1, Sally H. Cross1, Michelle M. Simon3, Paul K. Potter3, Nicholas M. Morton2 and Ian J. Jackson1,4,* ABSTRACT these diseases have a wide range of pathologies (Koopman et al., Isocitrate dehydrogenase (IDH) is an enzyme required for the 2012). Common to many of the diseases, however, are neurological production of α-ketoglutarate from isocitrate. IDH3 generates the or neuromuscular manifestations, including retinal disease. We have NADH used in the mitochondria for ATP production, and is a tetramer identified a missense mutation in the gene encoding a subunit of the made up of two α, one β and one γ subunit. Loss-of-function and tricarboxylic acid (TCA) cycle enzyme isocitrate dehydrogenase 3 missense mutations in both IDH3A and IDH3B have previously been (IDH3) as causing a retinal degeneration phenotype in mice. There implicated in families exhibiting retinal degeneration. Using mouse are three isocitrate dehydrogenase isozymes in mammals. IDH1 and models, we investigated the role of IDH3 in retinal disease and IDH2 are both homodimers that catalyse the decarboxylation of α mitochondrial function. We identified mice with late-onset retinal isocitrate to -ketoglutarate with the concomitant reduction of NADP degeneration in a screen of ageing mice carrying an ENU-induced to NADPH. IDH1 is cytoplasmic, whereas IDH2 is localised to the mutation, E229K, in Idh3a.