Download (7MB)
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Safety Data Sheet Material Name: TUNGSTEN HEXAFLUORIDE SDS ID: MAT24560
Safety Data Sheet Material Name: TUNGSTEN HEXAFLUORIDE SDS ID: MAT24560 Section 1 - PRODUCT AND COMPANY IDENTIFICATION Material Name TUNGSTEN HEXAFLUORIDE Synonyms MTG MSDS 85; TUNGSTEN FLUORIDE (WF6), (OC-6-11)-; WOLFRAM HEXAFLUORIDE; HEXAFLUOROTUNGSTEN; TUNGSTEN(6+) FLUORIDE; TUNGSTEN HEXAFLUORIDE (WF6); TUNGSTEN VI FLUORIDE; TUNGSTEN FLUORIDE; UN 2196; F6W Chemical Family Fluoride, inorganic, metal Product Use Industrial and Specialty Gas Applications. Restrictions on Use None known. Details of the supplier of the safety data sheet MATHESON TRI-GAS, INC. 3 Mountainview Road Warren, NJ 07059 General Information: 1-800-416-2505 Emergency #: 1-800-424-9300 (CHEMTREC) Outside the US: 703-527-3887 (Call collect) Section 2 - HAZARDS IDENTIFICATION Classification in accordance with paragraph (d) of 29 CFR 1910.1200. Gases Under Pressure - Liquefied gas Acute Toxicity - Inhalation - Gas - Category 2 Skin Corrosion/Irritation - Category 1 Serious Eye Damage/Eye Irritation - Category 1 Specific target organ toxicity - Repeated exposure - Category 1 GHS Label Elements Symbol(s) Signal Word Danger Hazard Statement(s) Contains gas under pressure; may explode if heated. Fatal if inhaled. Causes severe skin burns and eye damage. Causes damage to organs through prolonged or repeated exposure. (bones ) Precautionary Statement(s) Prevention Do not breathe gas. Use only outdoors or in a well-ventilated area. In case of inadequate ventilation wear respiratory protection. ____________________________________________________________ Page 1 of 9 Issue date: 2021-07-07 Revision 7.0 Print date: 2021-07-07 Safety Data Sheet Material Name: TUNGSTEN HEXAFLUORIDE SDS ID: MAT24560 Do not eat, drink or smoke when using this product. Wear protective gloves/protective clothing/eye protection/face protection. -

Tungsten Hexafluoride WF6 Safety Data Sheet SDS P4855
Tungsten hexafluoride Safety Data Sheet P-4855 This SDS conforms to U.S. Code of Federal Regulations 29 CFR 1910.1200, Hazard Communication. Date of issue: 01/01/1985 Revision date: 10/24/2016 Supersedes: 04/28/2015 SECTION: 1. Product and company identification 1.1. Product identifier Product form : Substance Name : Tungsten hexafluoride CAS No : 7783-82-6 Formula : WF6 Other means of identification : Tungsten hexafluoride 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Industrial use. Use as directed. 1.3. Details of the supplier of the safety data sheet Praxair, Inc. 10 Riverview Drive Danbury, CT 06810-6268 - USA T 1-800-772-9247 (1-800-PRAXAIR) - F 1-716-879-2146 www.praxair.com 1.4. Emergency telephone number Emergency number : Onsite Emergency: 1-800-645-4633 CHEMTREC, 24hr/day 7days/week — Within USA: 1-800-424-9300, Outside USA: 001-703-527-3887 (collect calls accepted, Contract 17729) SECTION 2: Hazard identification 2.1. Classification of the substance or mixture GHS-US classification Liquefied gas H280 Acute Tox. 2 (Inhalation: gas) H330 Skin Corr. 1A H314 Eye Dam. 1 H318 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS04 GHS05 GHS06 Signal word (GHS-US) : DANGER Hazard statements (GHS-US) : H280 - CONTAINS GAS UNDER PRESSURE; MAY EXPLODE IF HEATED H314 - CAUSES SEVERE SKIN BURNS AND EYE DAMAGE H330 - FATAL IF INHALED CGA-HG11 - SYMPTOMS MAY BE DELAYED CGA-HG22 - CORROSIVE TO THE RESPIRATORY TRACT Precautionary statements (GHS-US) : P202 -

Certain Data Contained in This Document May Be Difficult to Read in Microfiche Products
NOTICE CERTAIN DATA CONTAINED IN THIS DOCUMENT MAY BE DIFFICULT TO READ IN MICROFICHE PRODUCTS. dSa./ NREL/MP-451-4778B . UC Category': 270 • DE92 NRHL/MP--451-4778B DE92 04032.1 Safety Analysi%^.eport For the Use of Hal|ardous Production Materials in Photovoltaic Applications at the National Renewable Energy Laboratory Volume II: Appendices •.-ft '4^ oil, R.S, Crandall B»P. Nelson P.D. Moskowitz V.M. Fthenakis *N?S! National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 A Division of Midwest Research Institute Operated for the U.S. Department of Energv under Contract No. DE-AC02-83CH10093 " July 1992 Hi ^_ oismiuuiiuh or IMUI NOTICE Tnis report was ptepared as an account of work sponsoied by an agency of the United States government Neither the United Slates government nor any agency thereof nor any of their employees, makes any warranty express o< implied or assumes any legal liability Of tesponsibility'or the accuracy completeness or usefulness of any information apparatus product or process disclosed 01 represents that its use would not infringe privately owned, rights Reference herein to any specie commercial product process or service by trade name trademark manufacturer or otherwise ooes not necessarily constitute or imply its endorsement recommendation or favoring by the United Slates government or any agency lhereo> The views and opinions of authors expresseo herein do not necessarily state or reflect those of the United State; government or any agency theieof APPENDICES - TABLE OF CONTENTS A: AffCJtiftflt/RisK -

Gas and Metal Compatibility Guide
Gas & Metal Compatibility Table Chemical Suggested Choosing a Mott High Purity Gas Filter: Gas Formula Filter Media Choosing the best metal filter is not always a simple Ammonia NH 3 SS/Ni matter, because in addition to easily identified variables Argon Ar SS/Ni Arsenic Pentafluoride AsF 5 SS/Ni (i.e., gas, pressure and flow), there are subjective Arsine AsH 3 SS/Ni(1) considerations. Some gases are compatible with more Boron Trichloride BCI 3 Ni/H than one type of metal which allows you a choice when Boron Trifluoride BF 3 Ni/H Carbon Dioxide CO 2 SS/Ni selecting the right filter for your application. Carbon Monoxide * CO SS * Carbon Tetrachloride CCl 4 SS/Ni The information contained in this table is a guideline for Carbon Tetraflouride CF 4 SS/Ni appropriate filter selection. Consultation with your gas Chlorine Cl 2 SS/Ni/H Diborane B2H6 SS/Ni(1) supplier is recommended to ensure Dichlorosilane SiH 2Cl 2 Ni/H gas compatibility. Because so many Diethyltelluride C4H10 Te SS/Ni Fluorine F2 Ni/H factors can affect the chemical Freon 13 CClF 3 SS/Ni resistance of a given product, you Freon 14 Tetrafluoromethane CF 4 SS/Ni should pretest under your own Freon 23 Trifluoromethane/Fluoro-form CHF 3 SS/Ni Freon 115 Chloropentafluoroethane C2ClF 5 SS/Ni operating conditions. Freon 116 Hexafluoroethane C2F6 SS/Ni As with any Germane GeH 4 SS/Ni Helium He SS/Ni chemical Hydrogen H2 SS/Ni application, Hydrogen Bromide HBr Ni/H Hydrogen Chloride HCl Ni/H safety precautions Hydrogen Fluoride HF Ni/H as noted on MSDS Hydrogen Selenide H2Se SS/Ni sheets should be Hydrogen Sulfide H2S SS/Ni Krypton Kr SS/Ni observed . -

Comprehensive Electronics Solutions Praxair Offers a Full Suite of Electronics Gases, Equipment and Services
Comprehensive Electronics Solutions Praxair offers a full suite of electronics gases, equipment and services. Applications: Semiconductor From cost effectiveness to supply reliability to product quality – across North LEDs and Photonics America and around the world – Praxair is the gas supply partner with a proven Solar track record of success. Praxair’s experts are uniquely positioned to address the key needs of your operation, helping you remain competitive in today’s Nanotechnology highly complex, global electronics market. Electronics Assembly Product solutions with consistent performance Praxair is your industry-leading electronics solutions provider, backed by the dedicated service and industry know-how of the largest industrial gas company in North America. Whether its cylinder specialty gases for production processes, facility support gases from a microbulk system, or bulk or on-site delivery systems for large quantity demands, electronics manufacturing requires reliable, consistent products and service to get the job done right. Praxair’s complete suite of electronics gases, delivery equipment and services are designed to help you boost productivity and cut costs – crucial to your bottom line. Process gases that support your productivity Praxair’s global track record of supporting electronics producers in semiconductor, LED and electronics assembly markets among others is second to none. Our process gases maximize productivity, reduce process costs and enable new technologies. Count on Praxair for a reliable supply of gases that -

Low Temperature Chemical Vapor Deposition and Method for Depositing a Tungsten Silicide Film with Improved Uniformity and Reduced Fluorine Concentration
Europaisches Patentamt 19 European Patent Office Office europeen des brevets © Publication number : 0 591 086 A2 12 EUROPEAN PATENT APPLICATION © Application number : 93480132.5 © int. ci.5: H01L 21/3205, C23C 16/44, C23C 16/42, H01L 21/285 @ Date of filing : 10.09.93 The application is published incomplete as filed @ Inventor : Gow, Thomas Richard (Article 93 (2) EPC). The points in the 4 Lyman Drive description at which the omission obviously Williston, VT 05495 (US) occurs have been left blank. Inventor : Lebel, Richard John Box 2255A RD1 © Priority : 30.09.92 US 954783 Sheldon, VT 05483 (US) @ Date of publication of application : @ Representative : Klein, Daniel Jacques Henri 06.04.94 Bulletin 94/14 Compagnie IBM France Departement de Propriete Intellectuelle @ Designated Contracting States : F-06610 La Gaude (FR) DE FR GB © Applicant : International Business Machines Corporation Old Orchard Road Armonk, N.Y. 10504 (US) © Low temperature chemical vapor deposition and method for depositing a tungsten silicide film with improved uniformity and reduced fluorine concentration. © A tungsten silicide (WSix) film is formed by GROWTH RATE ( mg/m i n ) chemical vapor deposition using the reduction of tungsten hexafluoride (WF6) with dichlorosi- O — ro lane (SiH2CI2) referred to as DCS and a reduc- ing agent comprising silane (SiH4), hydrogen, or a mixture thereof. According to the method of the present invention, the WSix film is formed in a CVD reactor operating at a low pressure and at a wafer temperature between about 300 and 550°C. In the preferred embodiment, a thin nucleation layer is deposited before the WSix film. -

HIGH HAZARD GAS Review Date: 09/23/2019
University of Pittsburgh EH&S Guideline Number: 04-021 Safety Manual Subject: Effective Date: 04/19/2017 Page 1 of 9 HIGH HAZARD GAS Review Date: 09/23/2019 STORAGE AND USE OF HIGH HAZARD GAS 1. Definition of High Hazard (HH) Gases For these guidelines, any gas meeting one or more of the following definitions based on International Fire Code (IFC) and National Fire Protection Association (NFPA) standards: 1.1. Flammable gas – a material that is a gas at 68ºF (20ºC) or less at an absolute pressure of 14.7 psi (101.325 kPa) when in a mixture of 13% or less by volume with air, or that has a flammable range at an absolute pressure of 14.7 psi (101.325 kPa) with air of at least 12%, regardless of the lower limit 1.2. Pyrophoric gas – a gas with an autoignition temperature in air at or below 130ºF (54.4ºC) 1.3. Health Hazard 3 (HH3) gas – material that, under emergency conditions and according to the standards, can cause serious or permanent injury 1.4. Health Hazard 4 (HH4) gas – material that, under emergency conditions and according to the standards, can be lethal The storage and usage of a gas or gases meeting any of the above definitions must follow all applicable IFC and NFPA guidelines and the requirements outlined in this document. Consult EH&S for specific guidance on gas mixtures containing corrosive, flammable or poisonous gas components (ex. 1% carbon monoxide/nitrogen, 5% hydrogen sulfide/helium). 2. Notification Requirements Prior to Obtaining High Hazard Gases 2.1. -

AIGA 2005 Meeting
AIGA 2005 Meeting Best Practices in Compressed Gas Emergency Response Eugene Ngai Director of ER & Disposal Technology Air Products Singapore August 30, 2005 ElectronicElectronic SpecialtySpecialty MaterialsMaterials A typical Semiconductor Fab uses over 100 Electronic Specialty Materials (ESM) which include: Electronic Specialty Gases (ESG) Electronic Specialty Liquids (ESL) There are over 300 different Electronic Specialty Materials in common use. These have a wide variety of hazards, with many having multiple hazards. In some cases these are new materials which have limited data. These are supplied in a wide variety of packages ranging from small bubblers (50 gms) to large ISO containers (18,180 kg) Gas ER, Aug 2005 – E. Ngai AirAir ProductsProducts SuppliesSupplies ElectronicElectronic SpecialtySpecialty GasesGases whichwhich havehave aa VarietyVariety ofof HazardsHazards Ammonia Helium Perfluoropropane Argon Hydrogen Phosphine Arsine Hydrogen Bromide Phosphorus Pentafluoride Boron Trichloride Hydrogen Chloride Silane Boron Trifluoride Hydrogen Fluoride Silicon Tetrachloride Carbon Dioxide Hydrogen Iodide Silicon Tetrafluoride Carbon Tetrafluoride Hydrogen Selenide Sulfur Hexafluoride Chlorine Methyl Fluoride Sulfur Tetrafluoride Chlorine Trifluoride Methyltrichlorosilane Tetrafluoromethane Diborane Nitrogen Trichlorosilane Dichlorosilane Nitrogen Trifluoride Trimethylsilane Disilane Nitrous Oxide Tungsten Hexafluoride Fluorine Octafluorocyclobutane Xenon Halocarbon 23, 32, 116 Oxygen Gas ER, Aug 2005 – E. Ngai AirAir ProductsProducts -

Chemical Vapor Deposition of Tungsten Oxide Rein U
APPLIED ORGANOMETALLIC CHEMISTRY, VOL. 12, 155–160 (1998) Chemical Vapor Deposition of Tungsten Oxide Rein U. Kirss* and Lamartine Meda Department of Chemistry, Northeastern University, Boston, MA 02115, USA Crystalline and amorphous thin films of tung- literature contains several reports of electro-opti- sten(VI) oxide can be prepared by chemical cally inactive WO3 films prepared by sputtering, vapor deposition using a variety of volatile evaporation or spray methods,3 although later work precursors below 500 °C. Deposition parameters has yielded electrochromic WO3 films by these 1 for preparation of WO3 films from tungsten methods. No attempt is made here to review the hexacarbonyl [W(CO)6], tungsten hexafluoride literature pertaining to WO3 films prepared by these (WF6), tungsten ethoxides [W(OEt)x, x =5,6] methods. 3 and tetra(allyl)tungsten [W(h -C3H5)4] are sum- The present review focuses on CVD of amor- marized. The electrochromic behavior of these phous and crystalline tungsten oxide films, in- films is comparable with that observed for WO3 cluding thermal CVD, plasma-enhanced CVD films prepared by evaporation, sputtering and (PECVD) and photo-assisted CVD (PACVD). The electrodeposition. # 1998 John Wiley & Sons, principle advantage of chemical vapor deposition Ltd. of electronic materials over other methods is in step Appl. Organometal. Chem. 12, 155–160 (1998) coverage, the absence of radiation damage, throughput and the possibility for selective 4 Keywords: tungsten trioxide; electrochromism; growth. For example, CVD of WO3 occurs at thin films; chemical vapor deposition (CVD) temperatures significantly below those for evapora- ° 5 Received 5 December 1996; accepted 3 March 1997 tion (1300 C). -
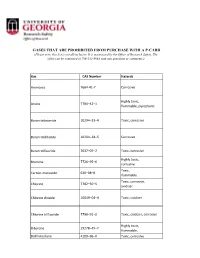
Lists of Gases for Pcard Manual.Xlsx
GASES THAT ARE PROHIBITED FROM PURCHASE WITH A P-CARD (Please note, this list is not all-inclusive. It is maintained by the Office of Research Safety. The office can be contacted at 706-542-9088 with any questions or comments.) Gas CAS Number Hazards Ammonia 7664‐41‐7 Corrosive Highly toxic, Arsine 7784–42–1 flammable, pyrophoric Boron tribromide 10294–33–4 Toxic, corrosive Boron trichloride 10294–34–5 Corrosive Boron trifluoride 7637–07–2 Toxic, corrosive Highly toxic, Bromine 7726–95–6 corrosive Toxic, Carbon monoxide 630–08–0 flammable Toxic, corrosive, Chlorine 7782–50–5 oxidizer Chlorine dioxide 10049–04–4 Toxic, oxidizer Chlorine trifluoride 7790–91–2 Toxic, oxidizer, corrosive Highly toxic, Diborane 19278–45–7 flammable, Dichlorosilane 4109–96–0 Toxic, corrosive Ethylene oxide 75–21–8 Toxic, flammable Highly toxic, corrosive, Fluorine 7782–41–4 oxidizer Highly toxic, Germane 7782–65–2 flammable Hydrogen bromide 10035–10–6 Toxic, corrosive Hydrogen chloride 7647–01–0 Toxic, corrosive Hydrogen cyanide 74–90–8 Highly toxic, flammable Hydrogen fluoride 7664–39–3 Toxic, corrosive Hydrogen iodide 10034‐85‐2 Toxic, corrosive Highly toxic, Hydrogen selenide 7783–07–5 flammable Toxic, flammable, Hydrogen sulfide 7783–06–4 corrosive Methyl bromide 74–83–9 Toxic Methyl isocyanate 624‐83‐9 Highly toxic, flammable Methyl mercaptan 74–93–1 Toxic, flammable Nickel carbonyl 13463–39–3 Highly toxic, flammable Highly toxic, Nitric oxide 10102–43–9 oxidizer Highly toxic, Nitrogen dioxide 10102–44–0 oxidizer, corrosive Highly toxic, Ozone 10028‐15‐6 -

Corrosive and Dangerous Gas Policy
Corrosive and Dangerous Gas Policy This Corrosive and Dangerous Gas Policy is applicable for models: SmartTrak 50, SmartTrak 100, SmartTrak 140, MicroTrak 101, MassTrak 810, TopTrak 820, and SideTrak 830/840 Series capillary products. This policy is intended to offer specific information regarding the service and warranty of Sierra’s 50, 100 and 800 Series capillary products in applications where the gas to be used can corrode stainless steel. This most often occurs with gases that, in the presence of air, become powerful acids. Usually, the result of removing an instrument from a proper installation. It is rarely the result of “wet” gas at your location or improper handling, though this is always a concern. Instead, it is because of the difficulty encountered in completely purging the reactive gas from the instrument. When such a device is returned to our facility (where humidity is always present), it becomes exposed to moisture. This begins the corrosion process. The simple nature of removing such an instrument and returning it to us can damage the device. Return Policy Concerning Scientific Units Exposed to Corrosive and/or Dangerous Gases Sierra’s 50, 100 and 800 Series capillary products are compatible with most gases due to their 316 stainless steel flow bodies and sensors, provided they are used with the recommended elastomeric seal material. However, these gases can often mix with other gases or liquids during use which can alter their chemical properties and create a corrosive or dangerous condition as noted above. Because of safety concerns, Sierra has adopted a “Do Not Return” policy for meters and controllers subjected to the following gases: Gas Name Gas Symbol Boron Trichloride BCl3 Chlorine CL2 Fluorine F2 Hydrogen Chloride HCL Hydrogen Fluoride HF Hydrogen Sulfide H2S Nitrogen Dioxide NO2 Tungsten Hexafluoride WF6 Sulfur Dioxide SO2 Uranium Hexafluoride UF6 Updated: 5/8/20 1 Sierra Policy Summary 1. -

Method for Preparing Gaseous Metallic Fluoride
Europaisches Patentamt 0 333 084 J European Patent Office (p) Publication number: A2 Office europeen des brevets EUROPEAN PATENT APPLICATION @ Application number: 89104364.8 @) lnt.Cl.4:C01B 9/08 © Date of filing: 11.03.89 Priority: 16.03.88 JP 60271/88 Applicant: MITSUI TOATSU CHEMICALS INC. 16.03.88 JP 60272/88 No. 2-5, Kasumigaseki 3-chome 16.03.88 JP 60273/88 Chiyoda-Ku Tokyo 100(JP) 16.03.88 JP 60274/88 11.11.88 JP 283749/88 Inventor: Harada, Isao, Mr. 5-7-2, Hikoshima Sakomachi Date of publication of application: Shimonoseki-shi Yamaguchi-ken(JP) 20.09.89 Bulletin 89/38 Inventor: Yoda, Yukihiro, Mr. 6-1-23, Hikoshima Sakomachi Designated Contracting States: Shimonoseki-shi Yamaguchi-ken(JP) DE FR GB IT Inventor: iwanaga, Naruyuki, Mr. 5-7-1, Hikoshima Sakomachi Shimonoseki-shi Yamaguchi-ken(JP) Inventor: Nishitsuji, Toshihiko, Mr. 5-9-6-505, Hikoshima Sakomachi Shimonoseki-shi Yamaguchi-ken(JP) Inventor: Kikkawa, Akio, Mr. M-408, 3-1, Shinakada Nishimachi Shimonoseki-shi Yamaguchi-ken(JP) Representative: Schiiler, Horst, Dr. Kaiserstrasse 69 D-6000 Frankfurt/Main 1(DE) © Method for preparing gaseous metallic fluoride. © The method for preparing a gaseous metallic fluoride is here disclosed which comprises reacting a metal or its oxide with a fluorine gas or nitrogen trifluoride gas, the aforesaid method being characterized by comprising the steps of mixing the metal or its oxide with a molding auxiliary comprising a solid metallic fluoride which does I not react with fluorine and nitrogen trifluoride; molding the resulting mixture under pressure; and contacting the [molded pieces with the fluorine gas or nitrogen trifluoride gas, while the molded pieces are heated.