NEVIRAPINE Extended-Release Tablets-Medication Guide and PI
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

35 Cyproterone Acetate and Ethinyl Estradiol Tablets 2 Mg/0
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION PrCYESTRA®-35 cyproterone acetate and ethinyl estradiol tablets 2 mg/0.035 mg THERAPEUTIC CLASSIFICATION Acne Therapy Paladin Labs Inc. Date of Preparation: 100 Alexis Nihon Blvd, Suite 600 January 17, 2019 St-Laurent, Quebec H4M 2P2 Version: 6.0 Control # 223341 _____________________________________________________________________________________________ CYESTRA-35 Product Monograph Page 1 of 48 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION ....................................................................... 3 SUMMARY PRODUCT INFORMATION ............................................................................................. 3 INDICATION AND CLINICAL USE ..................................................................................................... 3 CONTRAINDICATIONS ........................................................................................................................ 3 WARNINGS AND PRECAUTIONS ....................................................................................................... 4 ADVERSE REACTIONS ....................................................................................................................... 13 DRUG INTERACTIONS ....................................................................................................................... 16 DOSAGE AND ADMINISTRATION ................................................................................................ 20 OVERDOSAGE .................................................................................................................................... -

Revised 4/1/2021 GEORGIA MEDICAID FEE-FOR-SERVICE HIV
GEORGIA MEDICAID FEE-FOR-SERVICE HIV-AIDS PA SUMMARY Preferred (may not be all inclusive) Non-Preferred Abacavir generic Abacavir/lamivudine/zidovudine generic Abacavir/lamivudine generic Aptivus (tipranavir) Complera (emtricitabine/rilpivirine/tenofovir disoproxil Atazanavir capsules generic fumarate) Atripla (efavirenz/emtricitabine/tenofovir disoproxil Crixivan (indinavir) fumarate) Biktarvy (bictegravir/emtricitabine/tenofovir Delstrigo (doravirine/lamivudine/tenofovir disoproxil alafenamide) fumarate) Cimduo (lamivudine/tenofovir disoproxil fumarate) Fuzeon (enfuvirtide) Descovy (emtricitabine/tenofovir alafenamide) Intelence (etravirine) Dovato Invirase (saquinavir) Edurant (rilpivirine)* Lexiva (fosamprenavir) Efavirenz tablets generic Nevirapine extended-release generic Emtriva (emtricitabine) Norvir Powder (ritonavir) Epivir solution (lamivudine) Pifeltro (doravirine) Evotaz (atazanavir/cobicistat)* Reyataz Powder (atazanavir) Genvoya (elvitegravir/cobicistat/emtricitabine/ Ritonavir tablets generic tenofovir alafenamide) Isentress and Isentress HD (raltegravir)* Rukobia (fostemsavir) Juluca (dolutegravir/rilpivirine) Selzentry (maraviroc) Kaletra (lopinavir/ritonavir) Stavudine generic^ Stribild (elvitegravir/cobicistat/emtricitabine/ tenofovir Lamivudine generic disoproxil fumarate) Symfi (efavirenz 600 mg/lamivudine/tenofovir Lamivudine/zidovudine generic disoproxil fumarate) Symfi Lo (efavirenz 400 mg/lamivudine/tenofovir Nevirapine immediate-release tablets generic disoproxil fumarate) Norvir (ritonavir) Temixys (lamivudine/tenofovir -

Pharmacokinetic Interactions Between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance
life Review Pharmacokinetic Interactions between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance Laura Rombolà 1 , Damiana Scuteri 1,2 , Straface Marilisa 1, Chizuko Watanabe 3, Luigi Antonio Morrone 1, Giacinto Bagetta 1,2,* and Maria Tiziana Corasaniti 4 1 Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, Section of Preclinical and Translational Pharmacology, University of Calabria, 87036 Rende, Italy; [email protected] (L.R.); [email protected] (D.S.); [email protected] (S.M.); [email protected] (L.A.M.) 2 Pharmacotechnology Documentation and Transfer Unit, Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Rende, Italy 3 Department of Physiology and Anatomy, Tohoku Pharmaceutical University, 981-8558 Sendai, Japan; [email protected] 4 School of Hospital Pharmacy, University “Magna Graecia” of Catanzaro and Department of Health Sciences, University “Magna Graecia” of Catanzaro, 88100 Catanzaro, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-0984-493462 Received: 28 May 2020; Accepted: 30 June 2020; Published: 4 July 2020 Abstract: The therapeutic efficacy of a drug or its unexpected unwanted side effects may depend on the concurrent use of a medicinal plant. In particular, constituents in the medicinal plant extracts may influence drug bioavailability, metabolism and half-life, leading to drug toxicity or failure to obtain a therapeutic response. This narrative review focuses on clinical studies improving knowledge on the ability of selected herbal medicines to influence the pharmacokinetics of co-administered drugs. Moreover, in vitro studies are useful to anticipate potential herbal medicine-drug interactions. -

Indinavir Sulfate Capsule Merck & Co., Inc
CRIXIVAN - indinavir sulfate capsule Merck & Co., Inc. ---------- CRIXIVAN® (INDINAVIR SULFATE) CAPSULES DESCRIPTION CRIXIVAN1 (indinavir sulfate) is an inhibitor of the human immunodeficiency virus (HIV) protease. CRIXIVAN Capsules are formulated as a sulfate salt and are available for oral administration in strengths of 100, 200, 333, and 400 mg of indinavir (corresponding to 125, 250, 416.3, and 500 mg indinavir sulfate, respectively). Each capsule also contains the inactive ingredients anhydrous lactose and magnesium stearate. The capsule shell has the following inactive ingredients and dyes: gelatin, titanium dioxide, silicon dioxide and sodium lauryl sulfate. The chemical name for indinavir sulfate is [1(1S,2R),5(S)]-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H-inden-1-yl)-5-[2-[[(1,1 dimethylethyl)amino]carbonyl]-4-(3-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl)-D-erythro-pentonamide sulfate (1:1) salt. Indinavir sulfate has the following structural formula: Indinavir sulfate is a white to off-white, hygroscopic, crystalline powder with the molecular formula C36H47N5O4• H2SO4 and a molecular weight of 711.88. It is very soluble in water and in methanol. 1 Registered trademark of MERCK & CO., Inc. COPYRIGHT © 1996, 1997, 1998, 1999, 2004 MERCK & CO., Inc. All rights reserved MICROBIOLOGY Mechanism of Action HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Indinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. -

Recommendations for the Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection Table of Contents Table 1
Recommendations for the Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection Table of Contents Table 1. Outline of the Guidelines Development Process..........................................................................................................................1 Table 2. Rating Scheme for Recommendations........................................................................................................................................3 Table 3. Sample Schedule for Clinical and Laboratory Monitoring of Children Before and After Initiation of Combination Antiretroviral Therapy .................................................................................................................4 Table 4. Primary FDA-Approved Assays for Monitoring Viral Load D-8 Table 5. HIV Infection Stage Based on Age-Specific CD4 Count or Percentage ........................................................................................4 Table 6. HIV-Related Symptoms and Conditions ......................................................................................................................................5 Table 7. Antiretroviral Regimens Recommended for Initial Therapy for HIV Infection in Children ...........................................................................................................................................................................................7 Table 8. Advantages and Disadvantages of Antiretroviral Components Recommended for Initial Therapy in Children ............................................................................................................................................................10 -
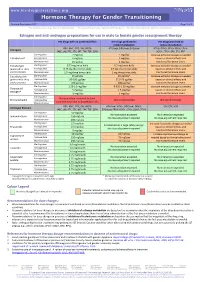
Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 for Personal Use Only
www.hiv-druginteractions.org Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 For personal use only. Not for distribution. For personal use only. Not for distribution. For personal use only. Not for distribution. Estrogen and anti-androgen preparations for use in male to female gender reassignment therapy HIV drugs with no predicted effect HIV drugs predicted to HIV drugs predicted to inhibit metabolism induce metabolism RPV, MVC, DTG, RAL, NRTIs ATV/cobi, DRV/cobi, EVG/cobi ATV/r, DRV/r, FPV/r, IDV/r, LPV/r, Estrogens (ABC, ddI, FTC, 3TC, d4T, TAF, TDF, ZDV) SQV/r, TPV/r, EFV, ETV, NVP Starting dose 2 mg/day 1 mg/day Increase estradiol dosage as needed Estradiol oral Average dose 4 mg/day 2 mg/day based on clinical effects and Maximum dose 8 mg/day 4 mg/day monitored hormone levels. Estradiol gel Starting dose 0.75 mg twice daily 0.5 mg twice daily Increase estradiol dosage as needed (preferred for >40 y Average dose 0.75 mg three times daily 0.5 mg three times daily based on clinical effects and and/or smokers) Maximum dose 1.5 mg three times daily 1 mg three times daily monitored hormone levels. Estradiol patch Starting dose 25 µg/day 25 µg/day* Increase estradiol dosage as needed (preferred for >40 y Average dose 50-100 µg/day 37.5-75 µg/day based on clinical effects and and/or smokers) Maximum dose 150 µg/day 100 µg/day monitored hormone levels. Starting dose 1.25-2.5 mg/day 0.625-1.25 mg/day Increase estradiol dosage as needed Conjugated Average dose 5 mg/day 2.5 mg/day based on clinical effects and estrogen† Maximum dose 10 mg/day 5 mg/day monitored hormone levels. -

Divestra Leaflet
• Known or suspected estrogen-dependent neoplasia Drugs which may decrease the therapeutic effect of Cyproterone acetate+Ethinyl Pregnancy and Breastfeeding • Undiagnosed abnormal vaginal bleeding Combination of Cyproterone acetate and Ethinyl estradiol is contraindicated during estradiol and increase the incidence of breakthrough bleeding • Any ocular lesion arising from ophthalmic vascular disease, such as partial or complete pregnancy and breastfeeding. loss of vision or defect in visual fields Effects on ability to drive and use machines • Concomitant use with other Estrogen+Progestogen combinations or estrogens or Unknown progestogens alone • When pregnancy is suspected or diagnosed Adverse Reactions • Severe diabetes with vascular changes Common adverse reactions includes headaches, nausea, abdominal pain, weight gain, • A history of otosclerosis with deterioration during pregnancy depressed or altered mood and breast pain or tenderness. • Hypersensitivity to this drug or to any ingredient in the formulation or component of the Uncommon adverse reactions include vomiting, diarrhea, fluid retention and migraine. container. Overdose (Cyproterone acetate + Warnings and Precautions There is no antidote and treatment should be symptomatic. Ethinyl estradiol) Discontinue Cyproterone acetate+Ethinyl estradiol tablets at the earliest manifestation of the following: PHARMACOLOGICAL PROPERTIES • Thromboembolic and Cardiovascular Disorders such as thrombophlebitis, pulmonary Pharmacotherapeutic group: Sex hormones and modulators of the genital system, QUALITATIVE AND QUANTITATIVE COMPOSITION embolism, cerebrovascular disorders, myocardial ischemia, mesenteric thrombosis, and anti-androgens and estrogens. ATC code: G03HB01 Each film-coated tablet contains: retinal thrombosis. Cyproterone acetate (Ph.Eur.)………...2 mg • Conditions that predispose to Venous Stasis and to Vascular Thrombosis (eg. Mechanism of action Ethinyl estradiol (USP)……………...0.035 mg immobilization after accidents or confinement to bed during long-term illness). -

Original Article Factors Influencing Efavirenz and Nevirapine Plasma Concentration: Effect of Ethnicity, Weight and Co-Medication
Antiviral Therapy 13:675–685 Original article Factors influencing efavirenz and nevirapine plasma concentration: effect of ethnicity, weight and co-medication Wolfgang Stöhr1, David Back 2, David Dunn1, Caroline Sabin3, Alan Winston4, Richard Gilson5, Deenan Pillay 6, Teresa Hill 3, Jonathan Ainsworth7, Anton Pozniak8, Clifford Leen9, Loveleen Bansi3, Martin Fisher10, Chloe Orkin11, Jane Anderson12, Margaret Johnson13, Phillippa Easterbrook14, Sara Gibbons2 and Saye Khoo2* on behalf of the Liverpool TDM Database and the UK CHIC Study 1MRC Clinical Trials Unit, London, UK 2University of Liverpool, Liverpool, UK 3Department of Primary Care and Population Sciences, Royal Free and University College Medical School, London, UK 4St. Mary’s Hospital, London, UK 5Mortimer Market Centre, Royal Free and University College Medical School (RFUCMS), London, UK 6Department of Infection, RFUCMS, Centre for Infection, Health Protection Agency, London, UK 7North Middlesex University Hospital, London, UK 8Chelsea and Westminster NHS Trust, London, UK 9University of Edinburgh, Western General Hospital, Edinburgh, UK 10Brighton and Sussex University Hospitals NHS Trust, Sussex, UK 11Barts and The London NHS Trust, London, UK 12Homerton Hospital, London, UK 13Royal Free NHS Trust and RFUCMS, London, UK 14King’s College Hospital, London, UK *Corresponding author: E-mail: [email protected] Background: The aim of this study was to examine and zidovudine (25% lower; P=0.010). Notably, without factors influencing plasma concentration of efavirenz and adjustment for other factors, patients on rifampicin had nevirapine. 48% higher efavirenz concentration, as these patients Methods: Data from the Liverpool Therapeutic Drug Mon- were mostly black and on 800 mg/day. For nevirapine the itoring (TDM) registry were linked with the UK Collabora- predictors were black ethnicity (39% higher; P=0.002), tive HIV Cohort (CHIC) Study. -

Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis
Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis Centers for Disease Control and Prevention Office of Infectious Diseases National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination June 2013 This document is accessible online at http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Suggested citation: CDC. Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis [online]. 2013. Available from URL: http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Table of Contents Introduction 1 Methodology for Preparation of these Guidelines 2 The Role of Rifamycins in Tuberculosis Treatment 4 Managing Drug Interactions with Antivirals and Rifampin 5 Managing Drug Interactions with Antivirals and Rifabutin 9 Treatment of Latent TB Infection with Rifampin or Rifapentine 10 Treating Pregnant Women with Tuberculosis and HIV Co-infection 10 Treating Children with HIV-associated Tuberculosis 12 Co-treatment of Multidrug-resistant Tuberculosis and HIV 14 Limitations of these Guidelines 14 HIV-TB Drug Interaction Guideline Development Group 15 References 17 Table 1a. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in adults 21 Table 1b. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in children 22 Table 2a. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in adults 23 Table 2b. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in children 25 Table 3. Recommendations for co-administering antiretroviral drugs with RIFABUTIN in adults 26 ii Introduction Worldwide, tuberculosis is the most common serious opportunistic infection among people with HIV infection. -

Fatal Nevirapine-Induced Stevens- Johnson Syndrome with HIV-Associated Mania
CASE REPORT Fatal nevirapine-induced Stevens- Johnson syndrome with HIV-associated mania Z Zingela,1 MB ChB, MMed (Psych), FC Psych (SA); A Bronkhorst,1 MB ChB, DMH (SA); W M Qwesha,2 MB ChB; B P Magigaba,2 MB ChB, FC Derm (SA) 1 Department of Psychiatry, Port Elizabeth Hospital Complex, Walter Sisulu University, South Africa 2 Department of Dermatology, Port Elizabeth Hospital Complex, Walter Sisulu University, South Africa Corresponding author: Z Zingela ([email protected]) Mania with psychotic features is one of the common presenting clusters of psychiatric symptoms in HIV-infected patients. Commonly, patients with HIV-associated mania receive antiretroviral treatment, mood stabilisers and antipsychotics. This case of Stevens-Johnson syndrome highlights the dilemmas and complications that may arise when prescribing multiple medications in HIV-associated psychiatric disorders. S Afr J HIV Med 2014;15(2):65-66. DOI:10.7196/SAJHIVMED.1011 HIV enters the central nervous system early status and CD4+ count, ethnic background, age and gender. in the course of HIV infection and causes a Affected individuals with SJS/TEN are genetically predisposed range of neuropsychiatric complications, inclu- to developing severe cutaneous reactions based on the major ding HIV encephalopathy, depression, ma- histocompatibility complex molecules on their leukocyte cell [1] [10] nia, cognitive disorders and frank demen tia. surface. A study in Taiwan showed that 100% of Han Chinese Mania is one of the most common psychiatric presentations in patients who developed SJS in response to carbamazepine HIV-infected patients and requires antiretroviral therapy (ART), had an HLA B*1502 allele.[11] Comorbidity of SJS/TEN and mood stabilisers and antipsychotics to increase patient quality of HIV is posing a challenge in sub-Saharan Africa due to the life and decrease mortality.[1-3] ART may also protect from further high prevalence of HIV. -

Indinavir PK Fact Sheet Reviewed March 2016 Page 1 of 2 for Personal Use Only
www.hiv-druginteractions.org Indinavir PK Fact Sheet Reviewed March 2016 Page 1 of 2 For personal use only. Not for distribution. For personal use only. Not for distribution. For personal use only. Not for distribution. Details Generic Name Indinavir Trade Name Crixivan® Class Protease Inhibitor Molecular Weight 711.88 (as sulphate) Structure HO OH N N H H SO N N 2 4 O HN O H3C CH3 H3C Summary of Key Pharmacokinetic Parameters Plasma half life 1.8 ± 0.4 h Cmax 8.97 ± 2.87 µg/ml (800 mg every 8 hours) Cmin 0.146 µg/ml (800 mg every 8 hours) AUC 21.82 ± 8.11 µg/ml.h (800 mg every 8 hours) Bioavailability Approx 65% Absorption Administration of indinavir with a meal high in calories, fat, and protein resulted in a blunted and reduced absorption with an approximate 80 % reduction in AUC and an 86 % reduction in Cmax. Administration with light meals (e.g., dry toast with jam or fruit conserve, apple juice, and coffee with skimmed or fat–free milk and sugar or corn flakes, skimmed or fat–free milk and sugar) resulted in plasma concentrations comparable to the corresponding fasted values. Protein Binding ~60% Volume of Distribution ~1.74 L/kg [1] CSF:Plasma ratio 0.14 [2] Semen:Plasma ratio 1.9 [2] Renal Clearance <20% as unchanged drug Renal Impairment Safety in patients with impaired renal function has not been studied; less than 20% of indinavir is excreted in the urine as unchanged drug or metabolites. Hepatic Impairment Safety and efficacy of indinavir has not been established in patients with significant underlying liver disorders and should be used with caution. -

Drug-Drug Interaction Between Protease Inhibitors and Statins and Proton Pump Inhibitors
Drug-drug interaction between Protease inhibitors and statins and Proton pump inhibitors Item Type text; Electronic Report Authors Orido, Charles; McKinnon, Samantha Publisher The University of Arizona. Rights Copyright © is held by the author. Download date 01/10/2021 01:48:07 Item License http://rightsstatements.org/vocab/InC/1.0/ Link to Item http://hdl.handle.net/10150/636245 Group 47 :Orido/Samantha 1 Drug-drug interaction between Protease inhibitors and statins and Proton pump inhibitors Course Title: PhPr 862 Date: April 3, 2019 Faculty Advisor: Dr. Dan Malone Students: Charles Orido, Samantha McKinnon Pharm.D. Candidates, Class of 2019 Group 47 :Orido/Samantha 2 Objective The purpose of this article is to provide a systematic review of the pharmacokinetic and clinical data on drug-drug interactions between protease inhibitors (PIs) and statins, atazanavir and proton pump inhibitors (PPIs)and their clinical relevance. Methods A literature search was performed using Medline, EMBASE and google scholar, abstracts from 1970 to 2019 of major conferences were searched and FDA drug information package inserts of the manufacturer of every currently available PI was looked at. All data was summarized and verified by at least two investigators. Results A total of 246 references were identified, 8 of which were studies of pharmacokinetic and pharmacodynamics interactions between simvastatin, lovastatin and protease inhibitors and an additional 7 articles that provided pharmacokinetic of proton pump inhibitors and Atazanavir. Conclusions Protease inhibitors increases the AUC and Cmax of simvastatin by approximately 500% and 517% respectively. Therefore, simvastatin and Lovastatin are not recommended for a co-administration with a protease inhibitor.