Mass Spectrometric Investigation of the Aging Processes of Ballpoint Ink for the Examination of Questioned Documents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tombow Introduces New Dual Brush Pen Colors
FOR IMMEDIATE RELEASE November 20, 2018 Tombow Introduces 12 New Dual Brush Pen Colors ATLANTA – Now you have a dozen new reasons to love Tombow Dual Brush Pens. For the first time in 29 years, the iconic arts and crafts brand has introduced new colors to its wildly popular Dual Brush Pen line of water-based art markers. The 12 new colors added to the Dual Brush Pen lineup take the full collection from 96 to 108 colors. Earlier this year Tombow released a new Marker Case with 108 slots, developed specifically to store the full range of Dual Brush Pens. “These new Dual Brush Pen colors provide a well-balanced color assortment for all types of art and design projects,” said Director of Marketing Cathy Daniel. “We looked at our previous color assortment and tried to fill gaps to give our artists the best possible spectrum to use in their art projects. Many of the colors are also based on current trends.” Dual Brush Pens were first introduced in 1984, with additional colors added in 1989. Since that time no new colors have been introduced to the line. These brush pens are popular among artists, designers, and illustrators for their ease of use and durability. One end of the Dual Brush Pen features a brush tip that acts like a paintbrush, giving thick or thin lines by changing the amount of pressure added to the pen. The other end features a hard, fine tip that allows the user to create consistent lines. The water-based ink allows the Dual Brush Pens to blend and layer. -

Certified Products List
THE ART & CREATIVE MATERIALS INSTITUTE, INC. Street Address: 1280 Main St., 2nd Floor Mailing Address: P.O. Box 479 Hanson, MA 02341 USA Tel. (781) 293-4100 Fax (781) 294-0808 www.acminet.org Certified Products List March 28, 2007 & ANSI Performance Standard Z356._X BUY PRODUCTS THAT BEAR THE ACMI SEALS Products Authorized to Bear the Seals of The Certification Program of THE ART & CREATIVE MATERIALS INSTITUTE, INC. Since 1940, The Art & Creative Materials Institute, Inc. (“ACMI”) has been evaluating and certifying art, craft, and other creative materials to ensure that they are properly labeled. This certification program is reviewed by ACMI’s Toxicological Advisory Board. Over the years, three certification seals had been developed: The CP (Certified Product) Seal, the AP (Approved Product) Seal, and the HL (Health Label) Seal. In 1998, ACMI made the decision to simplify its Seals and scale the number of Seals used down to two. Descriptions of these new Seals and the Seals they replace follow: New AP Seal: (replaces CP Non-Toxic, CP, AP Non-Toxic, AP, and HL (No Health Labeling Required). Products bearing the new AP (Approved Product) Seal of the Art & Creative Materials Institute, Inc. (ACMI) are certified in a program of toxicological evaluation by a medical expert to contain no materials in sufficient quantities to be toxic or injurious to humans or to cause acute or chronic health problems. These products are certified by ACMI to be labeled in accordance with the chronic hazard labeling standard, ASTM D 4236 and the U.S. Labeling of Hazardous NO HEALTH LABELING REQUIRED Art Materials Act (LHAMA) and there is no physical hazard as defined with 29 CFR Part 1910.1200 (c). -
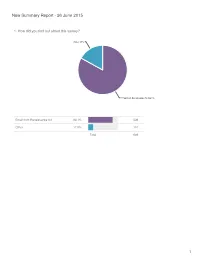
New Summary Report - 26 June 2015
New Summary Report - 26 June 2015 1. How did you find out about this survey? Other 17% Email from Renaissance Art 83.1% Email from Renaissance Art 83.1% 539 Other 17.0% 110 Total 649 1 2. Where are you from? Australia/New Zealand 3.2% Asia 3.7% Europe 7.9% North America 85.2% North America 85.2% 553 Europe 7.9% 51 Asia 3.7% 24 Australia/New Zealand 3.2% 21 Total 649 2 3. What is your age range? old fart like me 15.4% 21-30 22% 51-60 23.3% 31-40 16.8% 41-50 22.5% Statistics 21-30 22.0% 143 Sum 20,069.0 31-40 16.8% 109 Average 36.6 41-50 22.5% 146 StdDev 11.5 51-60 23.3% 151 Max 51.0 old fart like me 15.4% 100 Total 649 3 4. How many fountain pens are in your collection? 1-5 23.3% over 20 35.8% 6-10 23.9% 11-20 17.1% Statistics 1-5 23.3% 151 Sum 2,302.0 6-10 23.9% 155 Average 5.5 11-20 17.1% 111 StdDev 3.9 over 20 35.8% 232 Max 11.0 Total 649 4 5. How many pens do you usually keep inked? over 10 10.3% 7-10 12.6% 1-3 40.7% 4-6 36.4% Statistics 1-3 40.7% 264 Sum 1,782.0 4-6 36.4% 236 Average 3.1 7-10 12.6% 82 StdDev 2.1 over 10 10.3% 67 Max 7.0 Total 649 5 6. -

Materialien – Supplies
Materialien – Supplies Schulranzen/Backpacks Schulranzen Midi Plus Schulranzen Midi Plus Schulranzen Midi Plus Schulranzen Midi Plus Motorrad/schoolbag Formula 1/schoolbag Geometrie/schoolbag Heart/schoolbag Midi Midi Plus Motorcycle Midi Plus Formula 1 Midi Plus Geometry Plus Heart $ 95.70 $ 95.70 $ 95.70 $ 95.70 Schulranzen Midi Plus Schulranzen Midi Plus Schulranzen Midi Plus Schulranzen Midi Plus Race Car/schoolbag Rainbow Dinosaurier/schoolbag Horse/schoolbag Midi Midi Plus Race Car Butterfly/schoolbag Midi Midi Plus Dinosaur Plus Horse Plus Rainbow Butterfly $ 95.70 $ 95.70 $ 95.70 $ 95.70 Schulranzen Midi Plus Die Schulranzen enthalten alle auch ein gefülltes Unicorn/schoolbag Schüleretui (16 Teile), ein Federmäppchen und Midi Plus Unicorn einen Sportbeutel. $ 95.70 The schoolbags all come with a filled pencil case (16 pcs.), a pencil pouch, and a sports bag. Federmäppchen/Pencil Cases a b a b d c d c e e a. Federmäppchen Motorcross/pencil case a. Federmäppchen Fußball/pencil case soccer Motorcross $5.65 $5.65 b. Federmäppchen Race Car/pencil case race car b. Federmäppchen Driven/pencil case driven $5.65 $5.65 c. Federmäppchen Shark/pencil case shark $5.65 c. Federmäppchen Kick it/pencil case kick it $5.65 d. Federmäppchen Space/pencil case space $5.65 d. Federmäppchen Dino/pencil case dino $5.65 e. Federmäppchen Truck/pencil case truck $5.65 e. Federmäppchen Jet/pencil case jet $5.65 a b a b c c d d a. Federmäppchen Flower Heart/pencil case flower a. Federmäppchen Horse/pencil case horse $5.65 heart $5.65 b. Federmäppchen Rainbow Butterfly/pencil case b. -

Company UMB Bank, Statement Period 09/03/2019 to 10/01/2019
Eagle County School District Master Transaction Consolidation Transaction Search - Company UMB Bank, Statement Period 09/03/2019 to 10/01/2019 Mapped Cards Tran Date Account Company Unit Supplier Narrative Details Amount USD 9/4/2019 XXXX-XXXX-XXXX-1071 AVON ELEMENTARY Wal-Mart #1199 Purchase Wal-Mart #1199 4.35 9/6/2019 XXXX-XXXX-XXXX-1071 AVON ELEMENTARY Wm Supercenter #1199 Purchase Wm Supercenter #1199 17.58 9/15/2019 XXXX-XXXX-XXXX-1071 AVON ELEMENTARY Wal-Mart #1199 Purchase Wal-Mart #1199 20 9/4/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY Wm Supercenter #1199 comp books 3.5 9/7/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY Amzn Mktp Us physical education supplies girls on the run & instructional guide 84.9 9/9/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY Amzn Mktp Us 2nd grade headphones 79 9/9/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY Amzn Mktp Us comp. books for second grade 190.02 9/13/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY Hbs Online Dana Harrison Harvard training classes 399 9/21/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY Wm Supercenter #1199 break room supplies, coffee 71.84 9/22/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY The Home Depot #1525 break room supplies 14.2 9/24/2019 XXXX-XXXX-XXXX-0581 AVON ELEMENTARY United Deb Gregory conference DO reimbursement 258.6 9/3/2019 XXXX-XXXX-XXXX-1159 AVON ELEMENTARY Teacherspayteachers.Com Purchase Teacherspayteachers.Com reflections sheets 1.31 9/10/2019 XXXX-XXXX-XXXX-1159 AVON ELEMENTARY Ibarms IB ASSOC ANNUAL ROCKY MTN SCHOOLS 200 9/17/2019 XXXX-XXXX-XXXX-1159 AVON ELEMENTARY Teacherspayteachers.Com -

BBB-Summer-2018-Large.Pdf
‘Modern’ calligraphy has been making the rounds lately and if you're here — that's probably what you want to learn! But before we get all into the details, here's one thing you need to remember: calligraphy is not cursive. Cursive is joined up writing; you ideally write without li�ting your pen so you can write faster. While calligraphy can look similar, the key is to go slow and to break down each and every letter into its supporting strokes. THE BASIC STROKES ASCENDER LINE WAIST LINE BASE LINE DESCENDER LINE Or as some people call them, the drills! As dull as it sounds, brush lettering is made up of 9 common strokes illustrated above. With these strokes alone, you can form 21 out of the 26 letters in the lowercase alphabet — and some of the capital letters too! HOW THIS WORKS In Brush Basics, we'll be building your foundation right by covering all 9 strokes over the course of 2 weeks! Here's the schedule: On Day 1, you'll learn how to use your brush pen to form thin upstrokes and thick downstrokes. This is an easy lesson, so you have just one day to tackle it! Lesson 2 (delivered on Day 2) will cover underturn and overturn strokes. Lessons 3 and 4 (delivered on Days 4 and 6, respectively) will see you taking on the two di�ferent compound curves. You'll learn all about ovals in Lesson 5 (delivered on Day 8) — and because this is a hard one, you'll have not two but three days to practice it. -

Classification of Colouring / Writing Instruments and Related Art & Craft
European Writing Instrument Manufacturer’s Association Classification of Colouring / Writing Instruments and related Art & Craft Materials as Toys PRATERSTRASSE 34 · D-90429 NÜRNBERG · GERMANY Tel.: +49-(0) 911 / 27 229-0 · Fax +49-(0) 911 / 27 229-11 Email: [email protected] · Website: www.ewima.org European Writing Instrument Manufacturer’s Association 1 Products considered as „Toys“ - Chalks - Coloured pencils - Fibre pens - Finger paints - Drawing games (spiro games) - Modelling materials - Water colours - Wax crayons - Window colour - Fancy products [all products specially designed (e.g. by their shape) or clearly intended (e.g. by their packaging) for use in play by children under 14 years of age] “Toy”: Product designed or intended, whether or not exclusively, for use in play by children under 14 years of age. (As defined by European directive 2009/48/EC “safety of toys”, Art.2 No. 1). The final classification (product by product) and its inherent obligations, are the sole responsibility of each individual manufacturer, his authorized representative or importer. Products finally classified as toys by the manufacturer, his authorized representative or importer must comply with the essential safety requirements in Annex II of the directive or have successfully passed one of the conformity assessment procedures. The essential safety requirements are partially specified in the harmonized EN 71-series (all relevant parts). The products finally classified as toys must be marked with the CE-marking on the product itself or on the packaging. -

HCDE Procurement Card Report November Statement
HARRIS COUNTY DEPARTMENT OF EDUCATION December 2013 Disbursement Register - ACH Transfers Transaction Posting Date Payee/Description Amount Account 12/31/2013 TRS PAYMENT 294,282.53 General Operating & Payroll Clearing 12/13/2013 IRS PAYROLL TAXES 372,049.01 General Operating & Payroll Clearing 12/31/2013 IRS PAYROLL TAXES 387,819.93 General Operating & Payroll Clearing Total WIRE Transfers: $ 1,054,151.47 RECAP OF ALL DISBURSEMENTS Checks Total Printed PCard WIRES Disbursements Total General Operating & Payroll Clearing (100-199) $ 705,420.37 Total Special Revenue (200-400) 539,668.74 Total Internal Service/Facilities (700) 141,207.83 Total Fiduciary (800) 220.00 Total November PCard Activity Paid in December 49,086.04 Total ACH Transfers-Gen Operating & Payroll Clearing $ 1,054,151.47 $ 1,386,516.94 $ 49,086.04 $ 1,054,151.47 $ 2,489,754.45 Credit Card charges paid by check from above $ 3,901.91 (other than PCard) Harris County Department of Education Vendors with total aggregate payments of $50,000 or more in Fiscal Year 2014 as of December 31, 2013 Vendor Vendor Number Contract Type Sum of Payments BUTLER BUSINESS PRODUCTS 17320 JOB # 08/025JC 97,713.56 CBS PERSONNEL SERVICES LLC 61915 JOB # 13/001DG 50,548.29 CDW GOVERNMENT INC 18165 JOB #10/009LB 103,089.95 EDUCATION FOUNDATION OF HARRIS CTY 23957 Interlocal 190,000.00 EXECUTIVE THREAT SOLUTIONS LLC 85264 JOB # 12/007LB 184,758.34 GLAZIER FOODS COMPANY 27820 JOB # 13/039SC 74,235.19 HARRIS COUNTY APPRAISAL DISTRICT 29680 Per Texas Law/Linebarger 75,721.00 HIGH POINT SANITARY SOLUTIONS -

Sharpie 35003 - Permanent Marker Extra Fine Point Blue Dozen-SAN35003 Budget Friendly
Sharpie 35003 - Permanent Marker Extra Fine Point Blue Dozen-SAN35003 Budget Friendly I'm definitely discover Sharpie 35003 - Permanent Marker Extra Fine Point Blue Dozen-SAN35003, an excellent quality Office Product and School Supply at budget friendly total price and save shipping on the market right from trusted online sites. Compare it right now, you may not be dissatisfied. See Product Image | Check Updated Price Now | Customer Reviews In total, Sharpie 35003 - Permanent Marker Extra Fine Point Blue Dozen-SAN35003 is the high quality Office Product and School Supply and we're positively offer the item. The customer reviews will help you know a solid signal on the value for money and stability of products. It can be read every testimony by buyers which can find out more about his or her encounter. Most of the consumer reviews tell that this School Supply are usually top standard which is additionally reasonably-priced. If or when anyone looking to buy a brilliant Office Product at a good value, Sharpie 35003 - Permanent Marker Extra Fine Point Blue Dozen-SAN35003 may well be the just one. Where to Purchase Sharpie 35003 - Permanent Marker Extra Fine Point Blue Dozen-SAN35003 Quickly? If you need to get Office Product at low price, Amazon.com is best location with the low price, it's excellent for every body who are are generally need to buying on Sharpie 35003 - Permanent Marker Extra Fine Point Blue Dozen-SAN35003 is also fantastic Office Product and School Supply. However, specific features had produced dissatisfaction to users too, however this really had a little results on their overall review. -

Brands and Product Lines & Website Guide
plus Brands and Product Lines & Website Guide starts on page 56 Last Updated April 5, 2021 CONTACT [email protected] with questions, corrections, additions, updates Page 2 of 67 Product Guide Acetate Sheets Rolls Pads Grafix Jacquard Products / Rupert, Gibbon & Spider, Inc. MacPherson's SLS Arts Texas Art Supply/Art Supply Network Adhesives Alvin & Company Atlas Tape - Channeled Resources Grafix Grex Airbrush H. Schmincke & Co. GmbH & Co. KG HK Holbein, Inc Imagination International Jacquard Products / Rupert, Gibbon & Spider, Inc. Lineco MacPherson's Newell Brands SLS Arts Speedball Art Products Tombow Yasutomo Ziller's, LLC Advertising Art Materials Retailer Magazine Airbrush Equipment and Supplies Armadillo Art & Craft Grafix Grex Airbrush H. Schmincke & Co. GmbH & Co. KG HK Holbein, Inc Iwata-Medea Inc. Jacquard Products / Rupert, Gibbon & Spider, Inc. Page 3 of 67 MacPherson's SLS Arts SINOART Shanghai Co., Ltd Texas Art Supply/Art Supply Network Ziller's, LLC Albums Art and Photo Hahnemuhle USA Lineco MacPherson's SLS Arts Texas Art Supply/Art Supply Network Uchida of America Architectural Supplies ACCO UK. - ACCO Brands, Derwent Alumicolor Alvin & Company Grafix Jack Richeson & CO Inc. MacPherson's SINOART Shanghai Co., Ltd SLS Arts STAEDTLER-Mars Limited Studio Designs Texas Art Supply/Art Supply Network Tombow Artboard MultiMedia Aitoh Co. (WCG Group LLC, dba Aitoh Co.) Alvin & Company Crescent Cardboard, LLC Fredrix Canvas Grafix Heinz Jordan and Company Limited Hilltop Paper LLC Jack Richeson & CO Inc. Lineco Ranger Industries SINOART Shanghai Co., Ltd SLS Arts Texas Art Supply/Art Supply Network Block Printing ABIG GERMANY Armadillo Art & Craft Cranfield Colours Page 4 of 67 Educational Art and Craft Supplies Edward C Lyons Co. -

Town of Franklin Annual Report 2009
TOWN OF FRANKLIN 2009 ANNUAL R EPORT 1 2 3 TABLE OF C ONTENTS Animal Control………………………………………………………………….............................................….78 Assessors, Board of…………………………………………………………...........................………............162 Board of Appeals………………………….…………………………………………..…………................…….82 Zoning Board of Appeal Decisions for 2008…..............................................................................83 Building Inspection Department …………….………………......................……………………………..……86 Cable Television Advisory Committee ……………………………….....................…………….……………..88 Charles River Pollution Control District……………………………………………………………............……91 Conservation Commission.........................................................................................................................93 Cultural Council ........................................................................................................................................94 Design Review Committee....................................................................................................................... 94 Elected and Appointed Town Officials.........................................................................................................6 Facts on Franklin.............................................................................................................. Inside Back Cover Finance Committee....................................................................................................................................95 -

JUDGMENT of the COURT of FIRST INSTANCE (First Chamber) 12 January 1995 *
VIHO v COMMISSION JUDGMENT OF THE COURT OF FIRST INSTANCE (First Chamber) 12 January 1995 * In Case T-102/92, Viho Europe BV, a company incorporated under Netherlands law whose registered office is in Maastricht (Netherlands), represented by Werner Kleinmann, Rechtsan walt, Stuttgart, with an address for service in Luxembourg at the Chambers of Dupong et Associés, 14A Rue des Bains, applicant, v Commission of the European Communities, represented by Bernd Langeheine and Berend Jan Drijber, of its Legal Service, acting as Agents, assisted by H. J. Freund, Rechtsanwalt, Frankfurt am Main, with an address for service in Luxembourg at the office of Georgios Kremlis, of the Legal Service, Wagner Centre, Kirchberg, defendant, supported by * Language of the case: German. II-19 JUDGMENT OF 12.1. 1995 — CASE T-102/92 Parker Pen Ltd, a company incorporated under English law whose registered office is in Newhaven (United Kingdom), represented by Carla Hamburger, of the Amsterdam Bar, with an address for service in Luxembourg at the Chambers of Marc Loesch, 11 Rue Goethe, intervener, APPLICATION for the annulment of the Commission decision of 30 September 1992 rejecting the complaint of Viho Europe BV that Parker Pen Ltd and its sub sidiaries infringed Article 85(1) of the EEC Treaty (IV/32.725 — Viho/Parker Pen II), THE COURT OF FIRST INSTANCE OF THE EUROPEAN COMMUNITIES (First Chamber), composed of: R. Schintgen, President, R. Garcia-Valdecasas, H. Kirschner, B. Vesterdorf and C. W. Bellamy, Judges, Registrar: H. Jung, having regard to the written procedure and further to the hearing on 3 May 1994, gives the following II-20 VIHOv COMMISSION Judgment Facts and procedure 1 The applicant, Viho Europe BV (hereafter 'Viho'), a company incorporated under Netherlands law, markets office equipment on a wholesale basis and imports and exports that equipment.