Consequences of Divergence and Introgression for Speciation in Andean Cloud Forest Birds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Kinetic Effects of Temperature on Rates of Genetic Divergence and Speciation Andrew P
Kinetic effects of temperature on rates of genetic divergence and speciation Andrew P. Allen*†, James F. Gillooly‡, Van M. Savage§, and James H. Brown†¶ *National Center for Ecological Analysis and Synthesis, 735 State Street, Suite 300, Santa Barbara, CA 93101; ‡Department of Zoology, University of Florida, Gainesville, FL 32611; §Bauer Center for Genomics Research, Harvard University, Boston, MA 02138; and ¶Department of Biology, University of New Mexico, Albuquerque, NM 87131 Contributed by James H. Brown, May 2, 2006 Latitudinal gradients of biodiversity and macroevolutionary dy- dependence of mass-specific metabolic rate, B (J⅐secϪ1⅐gϪ1) namics are prominent yet poorly understood. We derive a model (12–14): that quantifies the role of kinetic energy in generating biodiver- Ϫ1/4 ϪE/kT ϪE/kT sity. The model predicts that rates of genetic divergence and B ϭ B͞M ϭ boM e ϭ Boe , [1] speciation are both governed by metabolic rate and therefore Ϫ1 show the same exponential temperature dependence (activation where B is individual metabolic rate (J sec ), M is body mass (g), ؋ 10؊19 J). Predictions are T is absolute temperature (K), Bo is a normalization parameter 1.602 ؍ energy of Ϸ0.65 eV; 1 eV Ϫ1 Ϫ1 supported by global datasets from planktonic foraminifera for independent of temperature (J⅐sec ⅐g ) that varies with body Ϫ1/4 rates of DNA evolution and speciation spanning 30 million years. size as Bo ϭ boM (12), and bo is a normalization parameter As predicted by the model, rates of speciation increase toward the independent of body size and temperature that varies among tropics even after controlling for the greater ocean coverage at taxonomic and functional groups (12, 17). -
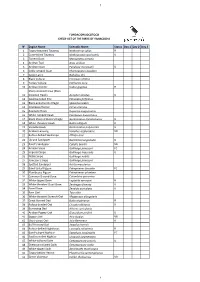
N° English Name Scientific Name Status Day 1
1 FUNDACIÓN JOCOTOCO CHECK-LIST OF THE BIRDS OF YANACOCHA N° English Name Scientific Name Status Day 1 Day 2 Day 3 1 Tawny-breasted Tinamou Nothocercus julius R 2 Curve-billed Tinamou Nothoprocta curvirostris U 3 Torrent Duck Merganetta armata 4 Andean Teal Anas andium 5 Andean Guan Penelope montagnii U 6 Sickle-winged Guan Chamaepetes goudotii 7 Cattle Egret Bubulcus ibis 8 Black Vulture Coragyps atratus 9 Turkey Vulture Cathartes aura 10 Andean Condor Vultur gryphus R Sharp-shinned Hawk (Plain- 11 breasted Hawk) Accipiter striatus U 12 Swallow-tailed Kite Elanoides forficatus 13 Black-and-chestnut Eagle Spizaetus isidori 14 Cinereous Harrier Circus cinereus 15 Roadside Hawk Rupornis magnirostris 16 White-rumped Hawk Parabuteo leucorrhous 17 Black-chested Buzzard-Eagle Geranoaetus melanoleucus U 18 White-throated Hawk Buteo albigula R 19 Variable Hawk Geranoaetus polyosoma U 20 Andean Lapwing Vanellus resplendens VR 21 Rufous-bellied Seedsnipe Attagis gayi 22 Upland Sandpiper Bartramia longicauda R 23 Baird's Sandpiper Calidris bairdii VR 24 Andean Snipe Gallinago jamesoni FC 25 Imperial Snipe Gallinago imperialis U 26 Noble Snipe Gallinago nobilis 27 Jameson's Snipe Gallinago jamesoni 28 Spotted Sandpiper Actitis macularius 29 Band-tailed Pigeon Patagoienas fasciata FC 30 Plumbeous Pigeon Patagioenas plumbea 31 Common Ground-Dove Columbina passerina 32 White-tipped Dove Leptotila verreauxi R 33 White-throated Quail-Dove Zentrygon frenata U 34 Eared Dove Zenaida auriculata U 35 Barn Owl Tyto alba 36 White-throated Screech-Owl Megascops -

UNIVERSIDAD COMPLUTENSE DE MADRID FACULTAD DE CIENCIAS BIOLÓGICAS Departamento De Zoología Y Antropología Física
UNIVERSIDAD COMPLUTENSE DE MADRID FACULTAD DE CIENCIAS BIOLÓGICAS Departamento de Zoología y Antropología Física TESIS DOCTORAL Diversidad y especificidad de simbiontes en aves neotropicales Diversity and host specificity of symbionts in neotropical birds MEMORIA PARA OPTAR AL GRADO DE DOCTOR PRESENTADA POR Michaël André Jean Moens Directores Javier Pérez Tris Laura Benítez Rico Madrid, 2017 © Michaël André Jean Moens, 2016 Diversidad y Especificidad de Simbiontes en Aves Neotropicales (Diversity and Host Specificity of Symbionts in Neotropical birds.) Tesis doctoral de: Michaël André Jean Moens Directores : Javier Pérez Tris Laura Benítez Rico Madrid, 2016 © Michaël André Jean Moens, 2016 Cover Front Chestnut-breasted Coronet (Boissoneaua matthewsii) Taken at the San Isidro Reserve, Ecuador. Copyright © Jaime Culebras Cover Back Royal Flycatcher (Onychorhynchus coronatus) Taken at the Nouragues Reserve, French Guiana. Copyright © Borja Milá UNIVERSIDAD COMPLUTENSE DE MADRID FACULTAD DE CIENCIAS BIOLÓGICAS DEPARTAMENTO DE ZOOLOGÍA Y ANTROPOLOGÍA FÍSICA Diversidad y Especificidad de Simbiontes en Aves Neotropicales (Diversity and Host Specificity of Symbionts in Neotropical birds.) Tesis doctoral de: Michaël André Jean Moens Directores: Javier Pérez Tris Laura Benítez Rico Madrid, 2016 © Michaël André Jean Moens, 2016 UNIVERSIDAD COMPLUTENSE DE MADRID FACULTAD DE CIENCIAS BIOLÓGICAS DEPARTAMENTO DE ZOOLOGÍA Y ANTROPOLOGÍA FÍSICA Diversidad y Especificidad de Simbiontes en Aves Neotropicales (Diversity and Host Specificity of Symbionts -

ECUADOR: the Andes Introtour and High Andes Extension 10Th- 19Th November 2019
Tropical Birding - Trip Report Ecuador: The Andes Introtour, November 2019 A Tropical Birding SET DEPARTURE tour ECUADOR: The Andes Introtour and High Andes Extension th th 10 - 19 November 2019 TOUR LEADER: Jose Illanes Report and photos by Jose Illanes Andean Condor from Antisana National Park This is one Tropical Birding’s most popular tours and I have guided it numerous times. It’s always fun and offers so many memorable birds. Ecuador is a wonderful country to visit with beautiful landscapes, rich culture, and many friendly people that you will meet along the way. Some of the highlights picked by the group were Andean Condor, White-throated Screech-Owl, Giant Antpitta, Jameson’s Snipe, Giant Hummingbird, Black-tipped Cotinga, Sword-billed Hummingbird, Club-winged Manakin, Lyre-tailed Nightjar, Lanceolated Monklet, Flame-faced Tanager, Toucan Barbet, Violet-tailed Sylph, Undulated Antpitta, Andean Gull, Blue-black Grassquit, and the attractive Blue-winged Mountain-Tanager. Our total species count on the trip (including the extension) was around 368 seen and 31 heard only. www.tropicalbirding.com +1-409-515-9110 [email protected] p.1 Tropical Birding - Trip Report Ecuador: The Andes Introtour, November 2019 Torrent Duck at Guango Lodge on the extension November 11: After having arrived in Quito the night before, we had our first birding this morning in the Yanacocha Reserve owned by the Jocotoco Foundation, which is not that far from Ecuador’s capital. Our first stop was along the entrance road near a water pumping station, where we started out by seeing Streak- throated Bush-Tyrant, Brown-backed Chat-Tyrant, Cinereous Conebill, White-throated Tyrannulet, a very responsive Superciliaried Hemispingus, Black-crested Warbler, and the striking Crimson-mantled Woodpecker. -

Allopatric Speciation with Little Niche Divergence Is Common Among
Journal of Biogeography (J. Biogeogr.) (2016) 43, 591–602 ORIGINAL Allopatric speciation with little niche ARTICLE divergence is common among alpine Primulaceae Florian C. Boucher1*, Niklaus E. Zimmermann2,3 and Elena Conti1 1Institute of Systematic Botany, University of ABSTRACT Zurich,€ 8008 Zurich,€ Switzerland, 2Dynamic Aim Despite the accumulation of cases describing fast radiations of alpine Macroecology, Swiss Federal Research plants, we still have limited understanding of the drivers of speciation in alpine Institute WSL, 8903 Birmensdorf, Switzerland, 3Department of Environmental floras and of the precise the timing of their diversification. Here, we investi- Systems Science, Swiss Federal Institute of gated spatial and temporal patterns of speciation in three groups of alpine Technology ETH, CH-8092 Zurich,€ Primulaceae. Switzerland Location Mountains of the European Alpine System. Methods We built a new phylogeny of Primulaceae including all species in three focal groups: Androsace sect. Aretia, Primula sect. Auricula and Soldanella. Combining phylogenetic information with a detailed climatic data set, we investigated patterns of range and ecological overlap between sister-species using an approach that takes phylogenetic uncertainty into account. Finally, we investigated temporal trajectories of diversification in the three focal groups. Results We found that a large majority of sister-species pairs in the three groups are strictly allopatric and show little differences in substrate and cli- matic preferences, a result that was robust to phylogenetic uncertainty. While rates of diversification have remained constant in Soldanella, both Androsace sect. Aretia and Primula sect. Auricula showed decreased diversification rates in the Pleistocene compared to previous geological epochs. Main conclusions Allopatric speciation with little niche divergence appears to have been by far the most common mode of speciation across the three groups studied. -

Creación De Un Manual Interpretativo Para El Buen
CARRERA ADMINISTRACIÓN TURÍSTICA Y HOTELERA CREACIÓN DE UN MANUAL INTERPRETATIVO SOBRE EL BUEN AVISTAMIENTO DE AVES EN LA RESERVA YANACOCHA UBICADA EN LA PARROQUIA DE NONO CANTÓN QUITO PROVINCIA PICHINCHA CON EL PROPÓSITO DE DAR A CONOCER EL AVITURISMO Proyecto de investigación previo a la obtención de título de tecnólogo en Administración Turística y Hotelera Autora: Amanda Estefania Tituaña Espinosa Tutor: Ing. Ximena Almeida Quito, Diciembre 2018 i Declaratoria Declaro que la investigación es absolutamente original, autentica, personal, que se han citado las fuentes correspondientes y en su ejecución se respetaron las disposiciones legales que protegen los derechos de autor vigentes. Las ideas, doctrinas resultados y conclusiones a los que he llegado son de mi absoluta responsabilidad. Amanda Estefania Tituaña Espinosa CC 1750804161 CREACIÓN DE UN MANUAL INTERPRETATIVO SOBRE EL BUEN AVISTAMIENTO DE AVES EN LA RESERVA YANACOCHA UBICADA EN LA PARROQUIA DE NONO CANTON QUITO PROVINCIA PICHINCHA CON EL PROPOSITO DE DAR A CONOCER EL AVITURISMO ii Licencia De Uso No Comercial Yo, Amanda Estefania Tituaña Espinosa portadora de la cedula de ciudadanía asignada Con el No. 175080416-1 de conformidad con lo establecido en el Artículo 110 del Código de Economía Social de los Conocimientos, la Creación y la Innovación (INGENIOS) que dice: “En el caso de las obras creadas en centros educativos,universidades,escuelas politécnicas, institutos superiores tecnicos,tecnólogos, pedagógicos, de arte y los conservatorios superiores , e institutos públicos de investigación como resultado de su actividad académica o de investigación tales como trabajos de titulación, proyectos de investigación o innovación, articulo académico , u otros análogos , sin perjuicio de que pueda existir relación de dependencia , la titularidad de los derechos patrimoniales corresponderá a los autores . -

Coupling, Reinforcement, and Speciation Roger Butlin, Carole Smadja
Coupling, Reinforcement, and Speciation Roger Butlin, Carole Smadja To cite this version: Roger Butlin, Carole Smadja. Coupling, Reinforcement, and Speciation. American Naturalist, Uni- versity of Chicago Press, 2018, 191 (2), pp.155-172. 10.1086/695136. hal-01945350 HAL Id: hal-01945350 https://hal.archives-ouvertes.fr/hal-01945350 Submitted on 5 Dec 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License vol. 191, no. 2 the american naturalist february 2018 Synthesis Coupling, Reinforcement, and Speciation Roger K. Butlin1,2,* and Carole M. Smadja1,3 1. Stellenbosch Institute for Advanced Study, Wallenberg Research Centre at Stellenbosch University, Stellenbosch 7600, South Africa; 2. Department of Animal and Plant Sciences, The University of Sheffield, Sheffield S10 2TN, United Kingdom; and Department of Marine Sciences, University of Gothenburg, Tjärnö SE-45296 Strömstad, Sweden; 3. Institut des Sciences de l’Evolution, Unité Mixte de Recherche 5554 (Centre National de la Recherche Scientifique–Institut de Recherche pour le Développement–École pratique des hautes études), Université de Montpellier, 34095 Montpellier, France Submitted March 15, 2017; Accepted August 28, 2017; Electronically published December 15, 2017 abstract: During the process of speciation, populations may di- Introduction verge for traits and at their underlying loci that contribute barriers Understanding how reproductive isolation evolves is key fl to gene ow. -

Selection in Finite Populations with Multiple Alleles
SELECTION IN FINITE POPULATIONS WITH MULTIPLE ALLELES. 111. GENETIC DIVERGENCE WITH CENTRIPETAL SELECTION AND MUTATION B. D. H. LATTER Division of Animal Genetics, C.S.I.R.O., Sydney, N.S.W., Australia Manuscript received April 28, 1971 Revised copy received December 6, 1971 ABSTRACT Natural selection for an intermediate level of gene or enzyme activity has been shown to lead to a high frequency of heterotic polymorphisms in popula- tions subject to mutation and random genetic drift. The model assumes a sym- metrical spectrum of mutational variation, with the majority of variants having only minor effects on the probability of survival. Each mutational event produces a variant which is novel to the population. Allelic effects are assumed to be additive on the scale of enzyme activity, heterosis arising whenever a heterozygote has a mean level of activity closer to optimal than that of other genotypes in the population.-A new measure of genetic divergence between populations is proposed, which is readily interpreted genetically, and increases approximately linearly with time under centripetal selection, drift and muta- tion. The parameter is closely related to the rate of accumulation of muta- tional changes in a cistron over an evolutionary time span.--A survey of published data concerning polymorphic loci in man and Drosophila suggests than an alternative model, based on the superiority of hybrid molecules, is not of general importance. Thirteen loci giving rise to hybrid zones on electrophor- esis have a mean heterozygote frequency of 0 22 rfr .OS, compared with a value of 0.23 i: .04 for 16 loci classified as producing no hybrid enzyme. -

Ecological Speciation in Phytophagous Insects
DOI: 10.1111/j.1570-7458.2009.00916.x MINI REVIEW Ecological speciation in phytophagous insects Kei W. Matsubayashi1, Issei Ohshima2 &PatrikNosil3,4* 1Department of Natural History Sciences, Hokkaido University, Sapporo 060-0810, Japan, 2Department of Evolutionary Biology, National Institute for Basic Biology, Okazaki 444-8585, Japan, 3Department of Ecology and Evolutionary Biology, University of Colorado, Boulder, CO 80309, USA, and 4Wissenschaftskolleg, Institute for Advanced Study, Berlin, 14193, Germany Accepted: 7 August 2009 Key words: host adaptation, genetics of speciation, natural selection, reproductive isolation, herbivo- rous insects Abstract Divergent natural selection has been shown to promote speciation in a wide range of taxa. For exam- ple, adaptation to different ecological environments, via divergent selection, can result in the evolution of reproductive incompatibility between populations. Phytophagous insects have been at the forefront of these investigations of ‘ecological speciation’ and it is clear that adaptation to differ- ent host plants can promote insect speciation. However, much remains unknown. For example, there is abundant variability in the extent to which divergent selection promotes speciation, the sources of divergent selection, the types of reproductive barriers involved, and the genetic basis of divergent adaptation. We review these factors here. Several findings emerge, including the observation that although numerous different sources of divergent selection and reproductive isolation can be involved in insect speciation, their order of evolution and relative importance are poorly understood. Another finding is that the genetic basis of host preference and performance can involve loci of major effect and opposing dominance, factors which might facilitate speciation in the face of gene flow. In addition, we raise a number of other recent issues relating to phytophagous insect speciation, such as alternatives to ecological speciation, the geography of speciation, and the molecular signatures of spe- ciation. -

Speciation in Geographical Setting
Speciation Speciation 2019 2019 The degree of reproductive isolation Substantial variation exists in among geographical sets of species - anagenesis populations within an actively 1859 1859 evolving species complex is often Achillea - yarrow tested by crossing experiments — as in the tidy tips of California 100K bp 100K bp back in time back in time back Rubus parviforus K = 4 mean population assignment 2 mya 2 mya ID CO WI_Door 5 mya BC_Hixton BC_MtRob WI_BruleS 5 mya BC_McLeod CA_Klamath MI_Windigo WA_Cascade WA_BridgeCr SD_BlackHills OR_Willamette MI_Drummond Speciation Speciation 2019 Reproductive isolation will ultimately stop all Although simple in concept, the recognition of species and thus the definition genetic connections among sets of populations of what are species have been controversial — more than likely due to the – cladogenesis or speciation continuum nature of the pattern resulting from the process of speciation 1859 Example: mechanical isolation via floral shape changes and pollinators between two parapatric species of California Salvia (sage) 100K bp back in time back 2 mya S. mellifera 5 mya Salvia apiana 1 Speciation Speciation Although simple in concept, the recognition of species and thus the definition Animal examples of speciation often show of what are species have been controversial — more than likely due to the clear reproductive barriers - hence zoologists continuum nature of the pattern resulting from the process of speciation preference (as opposed to botanists) for the Reproductive isolating Biological -

Ecological Speciation with Gene Flow in Neodiprion Pinetum and N. Lecontei
University of Kentucky UKnowledge Theses and Dissertations--Biology Biology 2020 FROM GENES TO SPECIES: ECOLOGICAL SPECIATION WITH GENE FLOW IN NEODIPRION PINETUM AND N. LECONTEI Emily E. Bendall University of Kentucky, [email protected] Author ORCID Identifier: https://orcid.org/0000-0003-2524-088X Digital Object Identifier: https://doi.org/10.13023/etd.2020.221 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Bendall, Emily E., "FROM GENES TO SPECIES: ECOLOGICAL SPECIATION WITH GENE FLOW IN NEODIPRION PINETUM AND N. LECONTEI" (2020). Theses and Dissertations--Biology. 62. https://uknowledge.uky.edu/biology_etds/62 This Doctoral Dissertation is brought to you for free and open access by the Biology at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Biology by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

Ecological and Life History Characteristics Predict Population Genetic Divergence of Two Salmonids in the Same Landscape
Molecular Ecology (2004) 13, 3675–3688 doi: 10.1111/j.1365-294X.2004.02365.x EcologicalBlackwell Publishing, Ltd. and life history characteristics predict population genetic divergence of two salmonids in the same landscape ANDREW R. WHITELEY, PAUL SPRUELL and FRED W. ALLENDORF Division of Biological Sciences, University of Montana, Missoula, MT 59812, USA Abstract Ecological and life history characteristics such as population size, dispersal pattern, and mating system mediate the influence of genetic drift and gene flow on population subdivision. Bull trout (Salvelinus confluentus) and mountain whitefish (Prosopium williamsoni) differ markedly in spawning location, population size and mating system. Based on these differences, we predicted that bull trout would have reduced genetic variation within and greater differ- entiation among populations compared with mountain whitefish. To test this hypothesis, we used microsatellite markers to determine patterns of genetic divergence for each species in the Clark Fork River, Montana, USA. As predicted, bull trout had a much greater propor- tion of genetic variation partitioned among populations than mountain whitefish. Among all sites, FST was seven times greater for bull trout (FST = 0.304 for bull trout, 0.042 for moun- tain whitefish. After removing genetically differentiated high mountain lake sites for each species FST, was 10 times greater for bull trout (FST = 0.176 for bull trout; FST = 0.018 for mountain whitefish). The same characteristics that affect dispersal patterns in these species also lead to predictions about the amount and scale of adaptive divergence among popula- tions. We provide a theoretical framework that incorporates variation in ecological and life history factors, neutral divergence, and adaptive divergence to interpret how neutral and adaptive divergence might be correlates of ecological and life history factors.