Brain Tumors : Practical Guide to Diagnosis and Treatment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Perceptual Attraction of Pre-Dominant Chords 1
Running Head: THE PERCEPTUAL ATTRACTION OF PRE-DOMINANT CHORDS 1 The Perceptual Attraction of Pre-Dominant Chords Jenine Brown1, Daphne Tan2, David John Baker3 1Peabody Institute of The Johns Hopkins University 2University of Toronto 3Goldsmiths, University of London [ACCEPTED AT MUSIC PERCEPTION IN APRIL 2021] Author Note Jenine Brown, Department of Music Theory, Peabody Institute of the Johns Hopkins University, Baltimore, MD, USA; Daphne Tan, Faculty of Music, University of Toronto, Toronto, ON, Canada; David John Baker, Department of Computing, Goldsmiths, University of London, London, United Kingdom. Corresponding Author: Jenine Brown, Peabody Institute of The John Hopkins University, 1 E. Mt. Vernon Pl., Baltimore, MD, 21202, [email protected] 1 THE PERCEPTUAL ATTRACTION OF PRE-DOMINANT CHORDS 2 Abstract Among the three primary tonal functions described in modern theory textbooks, the pre-dominant has the highest number of representative chords. We posit that one unifying feature of the pre-dominant function is its attraction to V, and the experiment reported here investigates factors that may contribute to this perception. Participants were junior/senior music majors, freshman music majors, and people from the general population recruited on Prolific.co. In each trial four Shepard-tone sounds in the key of C were presented: 1) the tonic note, 2) one of 31 different chords, 3) the dominant triad, and 4) the tonic note. Participants rated the strength of attraction between the second and third chords. Across all individuals, diatonic and chromatic pre-dominant chords were rated significantly higher than non-pre-dominant chords and bridge chords. Further, music theory training moderated this relationship, with individuals with more theory training rating pre-dominant chords as being more attracted to the dominant. -

The Strategic Half-Diminished Seventh Chord and the Emblematic Tristan Chord: a Survey from Beethoven to Berg
International Journal ofMusicology 4 . 1995 139 Mark DeVoto (Medford, Massachusetts) The Strategic Half-diminished Seventh Chord and The Emblematic Tristan Chord: A Survey from Beethoven to Berg Zusammenfassung: Der strategische halbverminderte Septakkord und der em blematische Tristan-Akkord von Beethoven bis Berg im Oberblick. Der halb verminderte Septakkord tauchte im 19. Jahrhundert als bedeutende eigen standige Hannonie und als Angelpunkt bei der chromatischen Modulation auf, bekam aber eine besondere symbolische Bedeutung durch seine Verwendung als Motiv in Wagners Tristan und Isolde. Seit der Premiere der Oper im Jahre 1865 lafit sich fast 100 Jahre lang die besondere Entfaltung des sogenannten Tristan-Akkords in dramatischen Werken veifolgen, die ihn als Emblem fUr Liebe und Tod verwenden. In Alban Bergs Lyrischer Suite und Lulu erreicht der Tristan-Akkord vielleicht seine hOchste emblematische Ausdruckskraft nach Wagner. If Wagner's Tristan und Isolde in general, and its Prelude in particular, have stood for more than a century as the defining work that liberated tonal chro maticism from its diatonic foundations of the century before it, then there is a particular focus within the entire chromatic conception that is so well known that it even has a name: the Tristan chord. This is the chord that occurs on the downbeat of the second measure of the opera. Considered enharmonically, tills chord is of course a familiar structure, described in many textbooks as a half diminished seventh chord. It is so called because it can be partitioned into a diminished triad and a minor triad; our example shows it in comparison with a minor seventh chord and an ordinary diminished seventh chord. -

MUS 211/211L Advanced Music Theory II and Lab MUS 313 Form and Analysis MUS 420
South Dakota State University MUS 211/211L Advanced Music Theory II and Lab MUS 313 Form and Analysis MUS 420 Concept: Music Theory "Compositional organization, such as pitch, including scale types and harmony; rhythm; texture; form; expressive elements, such as dynamics, articulation, tempo, and timbre; and basic aural skills: intervals, chords, scales, rhythms, melodies" MUS 211/211 L: Advanced Music Theory II and Lab (Note: since the skills and information regarding basic undergraduate music theory are taught in a "spiraling" manner through MUS 110, 111, 210, and 211, and MUS 211 is considered to be the course in which these materials are presented at a level commensurate with the level of the material as it appears on the Praxis test, this study guide reviews compositional organization as it was taught throughout the entire two-year theory sequence, and refers to pages in both the fall semester [MUS 210] and spring semester [MUS 211] class packets you purchased during the sophomore theory year.) Compositional Organization Students should refer to: The specific pages listed below from the class packets mentioned above, as well as Joe Dineen and Mark Bridge's Gig Bag Book of Theory and Harmony and Robert Ottman's Music for Sight Singing, 6th ed. (Note: the material and the page numbers in your class packets may vary slightly from what is listed below, depending on which year you took the course.) Specifically, students should review: From the fall semester of Sophomore Theory (MUS 210): o Rudiments (notation, scales/key, intervals, -
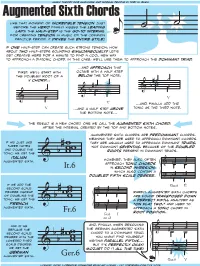
Augmented Sixth Chords Are Predominant Chords, Meaning They Are Used to Approach Dominant Chords
Augmentedmusic Sixth theory for musicians Chords and normal people by toby w. rush like that moment of incredible tension just before the hero finally kisses the leading lady, the half-step is the go-to interval for creating tension in music of the common ˙ practice period. it drives the entire style! ˙ if one half-step can create such strong tension, how about two half-steps sounding simultaneously? Let’s get creative here for a minute to find a cool new way to approach a diatonic chord. in this case, we’ll use them to approach the dominant triad. ...and approach that first, we’ll start with octave with a half step the doubled root of a below the top note, V chord... #˙ ˙ #˙ ˙ ˙ ˙ & b ˙ ˙ & b ˙ ˙ V & ˙ V ...and, finally, add the V ...and a half step above tonic as the third note. the bottom note... the result is a new chord, one we call the augmented sixth chord, after the interval created by the top and bottom notes. augmented sixth chords are predominant chords, meaning they are used to approach dominant chords. if we just use they are usually used to approach dominant triads, three notes not dominant sevenths, because of the doubled and double the roots present in dominant triads. tonic, we get the #w italian ww however, they also often augmented sixth. b w & approach tonic chords & #˙ ˙ It.6 in second inversion, ˙ ˙ which also contain a doubled fifth scale degree. ? b˙ n˙ b ˙ ˙ if we add the 6 Ger.6 I4 second scale degree instead ˙ ˙ rarely, augmented sixth chords of doubling the # w & ˙ ˙ are found transposed down tonic, we get the ww a perfect fifth, analyzed as french b w ˙ ˙ “on flat two,” and used to augmented sixth. -

The Augmented Sixth Chord
CHAPTER24 The Augmented Sixth Chord Characteristics, Derivation, and Behavior The two excerpts in Example 24.1 are from different style periods, yet they share several features. In terms of form and harmony, both divide into two subphrases and close with strong half cadences. Further, the pre-dominant harmony in both examples is the same: an altered iv6 chord. Indeed, we hear not a Phrygian cadence (iv6-V), but rather some chromatic version, where the diatonic major sixth above the bass is raised a half step to create the strongly directed interval of the augmented sixth (+6). The new half-step ascent (#4-5) mirrors the bass's half-step descent (6-5). We refer to such chromatic pre-dominants as augmented sixth chords because of the characteristic interval between the bass 6 and the upper-voice #4. Listen to both excerpts in Example 24.1, noting the striking sound of the augmented sixth chords. EXAMPLE 24.1 A. Schubert, WaltzinG minor, Die letzte Walzer, op. 127, no. 12, D. 146 472 CHAPTER 24 THE AUGMENTED SIXTH CHORD 473 B. Handel, "Since by Man Came Death," Messiah, HWV 56 Example 24.2 demonstrates the derivation of the augmented sixth chord from the Phrygian cadence. Example 24.2A represents a traditional Phrygian half cadence. In Example 24.2B, the chromatic F# fills the space between F and G, and the passing motion creates an interval of an augmented sixth. Finally, Example 24.2C shows the augmented sixth chord as a harmonic entity, with no consonant preparation. EXAMPLE 24.2 Phrygian Cadence Generates the Augmented Sixth Chord Given that the augmented sixth chord also occurs in major, one might ask if it is an example of an applied chord or a mixture chord? To answer this question, consider the diatonic progression in Example 24.3A. -

Describing Species
DESCRIBING SPECIES Practical Taxonomic Procedure for Biologists Judith E. Winston COLUMBIA UNIVERSITY PRESS NEW YORK Columbia University Press Publishers Since 1893 New York Chichester, West Sussex Copyright © 1999 Columbia University Press All rights reserved Library of Congress Cataloging-in-Publication Data © Winston, Judith E. Describing species : practical taxonomic procedure for biologists / Judith E. Winston, p. cm. Includes bibliographical references and index. ISBN 0-231-06824-7 (alk. paper)—0-231-06825-5 (pbk.: alk. paper) 1. Biology—Classification. 2. Species. I. Title. QH83.W57 1999 570'.1'2—dc21 99-14019 Casebound editions of Columbia University Press books are printed on permanent and durable acid-free paper. Printed in the United States of America c 10 98765432 p 10 98765432 The Far Side by Gary Larson "I'm one of those species they describe as 'awkward on land." Gary Larson cartoon celebrates species description, an important and still unfinished aspect of taxonomy. THE FAR SIDE © 1988 FARWORKS, INC. Used by permission. All rights reserved. Universal Press Syndicate DESCRIBING SPECIES For my daughter, Eliza, who has grown up (andput up) with this book Contents List of Illustrations xiii List of Tables xvii Preface xix Part One: Introduction 1 CHAPTER 1. INTRODUCTION 3 Describing the Living World 3 Why Is Species Description Necessary? 4 How New Species Are Described 8 Scope and Organization of This Book 12 The Pleasures of Systematics 14 Sources CHAPTER 2. BIOLOGICAL NOMENCLATURE 19 Humans as Taxonomists 19 Biological Nomenclature 21 Folk Taxonomy 23 Binomial Nomenclature 25 Development of Codes of Nomenclature 26 The Current Codes of Nomenclature 50 Future of the Codes 36 Sources 39 Part Two: Recognizing Species 41 CHAPTER 3. -

The Death and Resurrection of Function
THE DEATH AND RESURRECTION OF FUNCTION A Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By John Gabriel Miller, B.A., M.C.M., M.A. ***** The Ohio State University 2008 Doctoral Examination Committee: Approved by Dr. Gregory Proctor, Advisor Dr. Graeme Boone ________________________ Dr. Lora Gingerich Dobos Advisor Graduate Program in Music Copyright by John Gabriel Miller 2008 ABSTRACT Function is one of those words that everyone understands, yet everyone understands a little differently. Although the impact and pervasiveness of function in tonal theory today is undeniable, a single, unambiguous definition of the term has yet to be agreed upon. So many theorists—Daniel Harrison, Joel Lester, Eytan Agmon, Charles Smith, William Caplin, and Gregory Proctor, to name a few—have so many different nuanced understandings of function that it is nearly impossible for conversations on the subject to be completely understood by all parties. This is because function comprises at least four distinct aspects, which, when all called by the same name, function , create ambiguity, confusion, and contradiction. Part I of the dissertation first illuminates this ambiguity in the term function by giving a historical basis for four different aspects of function, three of which are traced to Riemann, and one of which is traced all the way back to Rameau. A solution to the problem of ambiguity is then proposed: the elimination of the term function . In place of function , four new terms—behavior , kinship , province , and quality —are invoked, each uniquely corresponding to one of the four aspects of function identified. -

Quasi Chords ? Glen Halls © All Rights Reserved
Quasi Chords ? Glen Halls © All Rights Reserved A quasi chord is a name given to functionally useful chords arrived at only by enharmonic adjustment or by correction. ( correction meaning when flats are resolved as sharps, basically) To put it another way, these are chords which are functionally useful and often familiar, despite their spelling. Quasi chords are typically simple structures which for all intents and purposes sound as triads and four note chords. The classic augmented sixth chord is one such structure. ( For example Ab+6 spelled, possibly, Ab C D F#, the 'French sixth' , and not Ab7 ) Less well documented are false triads in which the augmented second behaves as a minor third. It follows that many of these quasi chords are derived from inversions of diminished 7ths. A variation involves a suspension of some kind which resolves directly to tonic rather than first resolving internally relative to the root of this moving chord. Taking a quick look at the most common functional close tones relative to a C tonic: In the simplest possible sense quasi chords will involve any combination of at least two of the above dominants and subdominants , and typically one of each, which would be of use functionally but would not be spelled as a 1, 3, 5, or 7 of a tertian stack. As augmented sixths were mentioned earlier, let us quickly exhaust this set. Ab to F# (quasi Ab7) is one useful augmented sixth, as is Db to B, (quasi Db7- the so called 'tritone substitute' ) , and F to D#. (quasi F7 to Emi) There is one other possibility- the Fb to D resolution to C minor., or quasi E7 . -

An Exploration of Cultural Transmission Through the Application of Jazz Theory to the Music of Frederic Chopin
BearWorks MSU Graduate Theses Fall 2020 An Exploration of Cultural Transmission through the Application of Jazz Theory to the Music of Frederic Chopin Aaron Michael King Missouri State University, [email protected] As with any intellectual project, the content and views expressed in this thesis may be considered objectionable by some readers. However, this student-scholar’s work has been judged to have academic value by the student’s thesis committee members trained in the discipline. The content and views expressed in this thesis are those of the student-scholar and are not endorsed by Missouri State University, its Graduate College, or its employees. Follow this and additional works at: https://bearworks.missouristate.edu/theses Part of the Music Theory Commons Recommended Citation King, Aaron Michael, "An Exploration of Cultural Transmission through the Application of Jazz Theory to the Music of Frederic Chopin" (2020). MSU Graduate Theses. 3565. https://bearworks.missouristate.edu/theses/3565 This article or document was made available through BearWorks, the institutional repository of Missouri State University. The work contained in it may be protected by copyright and require permission of the copyright holder for reuse or redistribution. For more information, please contact [email protected]. AN EXPLORATION OF CULTURAL TRANSMISSION THROUGH THE APPLICATION OF JAZZ THEORY TO THE MUSIC OF FREDERIC CHOPIN A Master’s Thesis Presented to The Graduate College of Missouri State University TEMPLATE In Partial Fulfillment Of the Requirements for the Degree Master of Music By Aaron Michael King December 2020 Copyright 2020 by Aaron Michael King ii AN EXPLORATION OF CULTURAL TRANSMISSION THROUGH THE APPLICATION OF JAZZ THEORY TO THE MUSIC OF FREDERIC CHOPIN Music Missouri State University, December 2020 Master of Music Aaron Michael King ABSTRACT Connections between classical music and jazz were observed and detailed, providing an expanded understanding of the cultural underpinnings of Western music. -

Less Common Augmented Sixth Chord Spellings
MUS 357– Aaron Grant LESS COMMON AUGMENTED SIXTH CHORD SPELLINGS EXPLORE Although most of the augmented sixth chords you will find in the literature can be described as one of the three "national" types we have been working with, there are some less common variants of these chords that are important to discuss. Two of these types are displayed below. Analyze these passages with a partner, and discuss the two new forms of augmented sixth chords marked with arrows. How do they differ from typical augmented sixth chord spellings or voicings? Does the voice leading still work the same? Tchaikovsky, Sleeping Beauty, scene 1, mm. 62–78 MUS 357– Aaron Grant Schubert, "Der Doppelgänger," mm. 36–43 36 40 KEY POINTS In the Tchaikovsky excerpt, we see an example of a German diminished-third chord (Ger°3). This chord games its name because it places raised scale degree 4 in the bass, thus inverted the characteristic augmented sixth interval into a diminshed third. Note that: • This chord is typically approached through chromatic voice exchange. • The voice leading of a German diminished 7th chord still maintains the same voice leading as a typically German augmented sixth. In the Schubert song, we see an example of a secondary augmented-sixth chord—a type of chord that uses the same principals of the augmented sixth to lead to a tonic rather than a dominant chord. Note that: • The typical augmented sixth is maintained given ß^2 in the bass and 7^ in an upper voice. • This is an example of a German augmented sixth (albeit one that becomes a French), but any of our three main augmented sixth chords can appear as a secondary augmented sixth. -

The Augmented Sixth Chord
Theory Dr. Crist The Augmented Sixth Chord The augmented sixth chord was born in the Baroque period but occurs with greater frequency in the Romantic period. The aug. sixth chord may be understood as being derived from chromatic alterations of the iiº7 or iv7 chords. They do not function as subdominants, however, but function as predominants. All augmented sixth chords contain the interval of an augmented sixth, specifically scale degrees b6 and #4 which frame the dominant. The interval of the augmented sixth resolves outwards to an octave. The tonic also appears in every augmented sixth chord. There are three different "flavors" of augmented sixth chords: Italian, French, and German. They each include the augmented sixth, the tonic, and one other note as follows: Italian : double the tonic French - add the supertonic German : add the mediant. c minor: It+6 Fr+6 Gr+6 Progression: To avoid parallel fifths, the Gr+6 should resolve to iP before progressing to V. It+6 ---------- V or iP Fr+6 ---------- V or iP Gr+6 ---------- iP then to V c minor: It+6 Fr+6 Gr+6 Resolution: The interval of the augmented sixth resolves outwards. Common tones and stepwise motion should be used in the remaining voices. When the following chord is a V7, #4 should progress to natural 4 (the 7th of the V7): c minor: It+6 V7 The Individual Augmented Sixth Chords Italian - Derived from the progression i --- iv6 --- V. In order to intensify or embellish the melodic/harmonic progression to V, the root is altered and raised up a half-step creating a leading-tone relation to the dominant. -

ONLINE CHAPTER 2 CHROMATIC CHORDS FUNCTIONING AS the PREDOMINANT Artist in Residence: Jacqueline Du Pré
ONLINE CHAPTER 2 CHROMATIC CHORDS FUNCTIONING AS THE PREDOMINANT Artist in Residence: Jacqueline du Pré • Demonstrate how the Neapolitan and augmented sixth chords function as predominant • Notate Neapolitan and augmented sixth chords in various keys • Recognize Neapolitan and augmented sixth chords in musical context • Resolve Neapolitan and augmented sixth chords using proper voice leading Chapter Objectives NEAPOLITAN CHORDS In Chapter 8, we identified the importance of the relationship between tonic, subdominant, and dominant. Many of the progressions we have studied follow this movement throughout the entire composition. Study the following progression, and play it several times on your guitar or keyboard. G minor: i VI iv V i Both the tonic and submediant chords function as tonic, followed by the subdominant and the dominant chord leading to the cadence. Chromatic Chords Functioning as the Predominant |OL2-1 As we have seen in previous chapters, the use of chromaticism adds color, tension, and dissonance to a tonal composition. Using the previous progression, let’s add a few altered pitches to the subdominant chord. Instead of fa-le-do (4, ♭6, 1), how would the sound change if the do was raised and spelled enharmonically as a ra? What quality of chord would be created? Using the same progression above, realize both line 1 and line 2 on your guitar or keyboard. Gmin: i VI iv V i G- E ! C- D G- Gmin: i VI N6 V i G- E ! A !/C D G- Notice how the A♭ major chord is functioning as a predominant chord, leading to the D. The same principle works in major keys.