Genetic Analyses Reveal Demographic Decline and Population
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Navegaon (NN) Corridor, Gondia District of Maharashtra State, India
IJISET - International Journal of Innovative Science, Engineering & Technology, Vol. 8 Issue 5, May 2021 ISSN (Online) 2348 – 7968 | Impact Factor (2020) – 6.72 www.ijiset.com Ichthyofaunal Diversity and Conservation Perspective of Some Selected Wetlands in Nagzira – Navegaon (NN) Corridor, Gondia District of Maharashtra State, India Mahendra Bhojram Raut¹, Chintaman J. Khune², Laxman P. Nagpurkar³ ¹Research Scholar, J. M. Patel College Bhandara ([email protected])U30T ²Asso. Prof. & Head, P. G. Department of Zoology, M. B. Patel College Sakoli ([email protected] )U30T ³Asso. Prof. & Head, Department of Chemistry, M. B. Patel College Sakoli ([email protected] )U30T Abstract Ichthyofaunal diversity of five lakes in Nagzira Navegaon Corridor in Gondia district of Maharashtra, India was conducted to assess the fish fauna. The present investigation deals with the ichthyofaunal diversity in NN corridor during the year October 2014 to September 2016. The results of present study reveal the occurrence of 62 fish species belonging to different 18 families. Among the 18 families, Cyprinidae family dominated over remaining families, The Cyprinidae were reported 27 species followed by Bagridae with 5 species and Channidae with 4 species whereas 3 species each of Ambasiidae, Clariidae, Mastacembelidae, Siluridae and a single species was reported of family Anguillidae, Anabantidae, Belonidae, Gobiidae, Heteropneustidae, Osphrinemidae, Nandidae, Noteopteridae and Sisoridae. In the present investigation, the number of fish species recorded from each wetland vary from 34 to 51 species, it was 34 species in Putli lake, 35 species in Naktya lake, 45 species in Umarzari lake, 51 species in Rengepar lake and 38 species in Chulbandh lake. Majority of the fish 81% of the total number of species were classed as least concern followed by 10 % near threatened, 5% data deficient, 3% vulnerable and 1% endangered as per the IUCN red list categories observed during study period in NN corridor. -

Sustaining the Lives of Tigers and People
Annual Report, April 2014 Satpuda Landscape Tiger Programme Sustaining the Lives of Tigers and People Executive Summary THE TIGER CRISES The wild tiger is an iconic species, revered and feared With as few as 3,000 wild tigers left in the world, and in equal measure. Yet man’s fascination with the tiger numbers rapidly decreasing, the future for this iconic has not protected it from a mounting raft of threats species in its natural habitat is precarious indeed. In India, that have left as few as 3,000 worldwide clinging to home to more wild tigers than any other range country, survival. The Saptuda Highlands of Central India only 11% of original habitat remains in an increasingly arguably represent the best chance wild tigers fragmented and often degraded state. Whilst there are have for survival and here, the partners of the encouraging signs that the species might be on the rise Satpuda Landscape Tiger Programme (SLTP), in some areas, India could have as few as 1,400 tigers funded by the Born Free Foundation, are working remaining, requiring urgent protection to ensure any tirelessly to stem the tiger’s decline and aid its recovery can be sustained. recovery. Throughout 2013/14, the SLTP has maintained this dedication with a range of activities As a conservation dependent species, tigers require large detailed further in the report that follows, including: contiguous forests with access to water and undisturbed legal representation; landscape monitoring and core areas in which to breed. Against a backdrop of a lobbying; field research; mitigation of human- Tomorrow’s tiger conservationists! ©BNHS tiger conflict; health care provision; environmental burgeoning human population desperate to overcome forest resources, often resulting in fatalities on both sides, education programmes; and sustainable livelihood poverty, habitat is encroached upon for livestock grazing and it is clear that the threats to tigers are greater now than initiatives. -

NAWEGAON-NAGZIRA TIGER RESERVE Nawegaon-Nagzira Tiger Reserve Is Situated in Gondia and Shandara Districts of Maharashtra
NAWEGAON-NAGZIRA TIGER RESERVE Nawegaon-Nagzira Tiger Reserve is situated in Gondia and Shandara Districts of Maharashtra. The reserve is rich in bio-diversity and has linkages with Kanha, Pench and Tadoba Tiger Reserves. The topography is undulating, and the highest point viz. ‘Zenda Pahad’ is around 702 m above MSL. Area of the tiger reserve Core / Critical Tiger Habitat : 653.67 sq.km. Buffer Area : Yet to be constituted. The area details of the core / critical tiger habitats are as below: Sr. No. Name of Wildlife Sanctuary/National Park Area of Tiger Reserve in Sq.Km. 1 Nawegaon National Park 129.55 2 Nagzira Wildlife sanctuary 152.41 3 Nawegaon Wildlife Sanctuary 122.76 4 New Nagzira Wilidlife Sanctuary 151.33 5 Koka Wildlife Sanctuary 97.62 Total Area 653.67 Location Name of W.L.S./ N.P. Longitude Latitude 1.Nawegaon National Park 80° 5' to 80° 15' E 20° 45' to 21u 2' N 2.Nawegaon Wildlife Sanctuary 800 09' to 80u 21' E 20u 54' to 210 OS' N 3.Nagzira Wildlife Sanctuary 79058' to 80011' E 21 o 1 2' to 21 0 21' N 4.New Nagzira W.L.S. 79° 49' to 80u 11' E 21° 09' to 21° 20' N 5.Koka Wildlife Sanctuary 79° 43' to 79° 54' E 21° 06' to 21° 12' N 1 Habitat Attributes Biogeographically the reserve falls in the biotic province 6B: Biogeographic Kingdom : Paleotropical Sub - kingdom : Indomalayan Biogeographic zone : 6 -Deccan peninsula Biotic province : 6 - B - Central Deccan Flora The forests are "Southern Tropical Dry Deciduous" (Champion and Seth, 1968). -
Untamed Nagzira - OLD to BE DELETED
Untamed Nagzira - OLD TO BE DELETED About This Experience Nagzira is the dream nature gods dreamt! Nestled between Bhandara and Gondia districts, Nagzira is aptly called a green oasis in the middle of a dry and arid landscape of eastern Maharashtra. The jungle, dedicated to, and named aer Naagdev, oers great oppounities to observe and photograph spectacular wildlife including some of the top predators like the tiger, leopard and wild dogs. Highlights – • Nagzira gets its name from the god Naagdev, the guardian of the forest, and Pongezara, the eternal water source that originates from the hillock of Naagdev temple. • One of the very few places where one gets to stay in the core zone of the tiger reserve. • One of the best places in central India to observe leopards - a rarity in rest of the jungles. • The famous tiger named Jai was born and brought up in the Nagzira Tiger Reserve. • Although Vidarbha is known to be a dry region, Nagzira is covered in lush greenery throughout the year. The Footloose Factor – We at Footloose Journeys believe in creating value for our campers by taking them on meaningful travel experiences. Along with conducting wildlife safaris and wildlife photography sessions on the camps, we also make sure that our campers learn about the ecosystem as a whole. Through various discussion sessions, documentary screenings, quizzes, interactions with the members of tribal communities, and even interactions with forest depament personnel, we make sure that our campers take with them a bagful of beautiful memories and also deep knowledge about the place. Highlights To give you a more hands-on, rustic experience, we will spend quality time with the locals living in villages close to the sanctuary, geing to know the niy-griies of their lives. -

Navegaon-Nagzira Tiger Reserve: Maharashtra
Navegaon-Nagzira Tiger Reserve: Maharashtra drishtiias.com/printpdf/navegaon-nagzira-tiger-reserve-maharashtra Why in News Three labourers were killed and two others injured during an operation to douse a forest fire at Navegaon-Nagzira Tiger Reserve (NNTR) in Maharashtra. Key Points Location: It is situated in Gondia and Bhandara districts of Maharashtra. Gondia District shares common boundaries with the state of Madhya Pradesh and Chhattisgarh in the north and eastern side respectively. Strategically, the Tiger Reserve is located in the heart of central Indian Tiger landscape which contributes almost one sixth of the total tiger population of the country. Formation: It was notified as the 46th Tiger Reserve of India in December 2013. NNTR comprised of the notified area of Nawegaon National Park, Nawegaon Wildlife Sanctuary, Nagzira Wildlife Sanctuary, New Nagzira Wildlife Sanctuary and Koka Wildlife Sanctuary. Connectivity: NNTR has connectivity with the major tiger reserves in Central India like, Kanha and Pench tiger reserve in Madhya Pradesh, Tadoba-Andhari Tiger reserve in Maharashtra, Indravati Tiger Reserve in Chhattisgarh, Indirectly with the Kawal and Nagarjuna Sagar in Telangana and Andhra Pradesh and, Achanakmar Tiger reserve in Chhattisgarh. It is also connected to important tiger bearing areas like Umred-Karhandla sanctuary and Brahampuri Division (Maharashtra). 1/3 Flora: The major forest type is "Southern Tropical Dry Deciduous Forest". Few thorny plants are also found. Bamboo occurs in abundance. Fauna: Large Carnivores such as leopards and smaller carnivores like wild dogs, wolf jackals, jungle cats and also the good population of sloth bears are seen. The important herbivore includes Cheetal, Sambar, Nilgai, Chousingha, Barking deer, Wild pig and Indian gaur. -

To Tadoba with Love
SPECIAL FEATURE TO TADOBA WITH LOVE #NeverStopDiscovering—that attitude alone has led wildlife champions Poonam and Harshwardhan Dhanwatey on a lifelong journey to protect the forests where the tiger makes its home. From the safety of a Land Rover Discovery Sport we get the insider’s tour. By Prasad Ramamurthy. Photographs by Arjun Menon SPECIAL FEATURE Poonam and Harshwardhan Dhanwatey in a Land Rover Discovery Sport, at their private conservancy near the Tadoba-Andhari Tiger Reserve SPECIAL FEATURE Poonam and Harshwardhan Dhanwatey, founders of the NGO Tiger Research and Conservation Trust (TRACT). Clockwise from above: TRACT staffers looking at photos of animals spotted at Tigress@Ghosri; a tiger inside the reserve; prayers being offered to a tiger idol; the TRACT team with local volunteers n earthen lamp sits to one side of the dead person’s village.” There are at TRACT, also drawn from the local area, of a small tiger statue erected dozens of villages within the buffer zone teaches locals how to live with predators Aby the road. A bunch of incense that surrounds the reserve and predators, – the dos and don’ts of venturing into sticks circumambulate the idol. The ever so often, do lift livestock from these the forest; how to deal with a wild cat if it hands holding them belong to a dhoti- villages. People, too, have lost lives. And enters a village; what to do in the event of clad man who appears to be praying so, such belief is only to be expected a forest fire; how to report any suspicious fervently. -
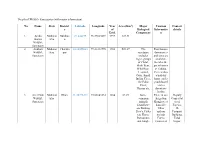
Details of Wildlife Sanctuaries (With Major Information) No. Name State District S Latitude Longitude Year of Estd. Area (Km2)
Details of Wildlife Sanctuaries (with major information) No. Name State District Latitude Longitude Year Area (Km2) Major Tourism Contact s of Biological Informatio details Estd. Component n 1. Amba Maharas Buldhan 21.244275 76.59823889 1997 127.11 Barwa htra a Wildlife Sanctuary 2. Andhari Maharas Chandra 20.16055833 79.41212778 1986 509.27 The Rest houses, Wildlife htra pur sanctuary dormitories Sanctuary includes and tents are Tiger, groups available. of Chital, Besides the Sloth Bear, guest houses Wild Boar, at Tadoba, Leopard, there is also Gaur, Small a holiday Indian Civet, home and a the Palm youth hostel Civet, with a Hyenas etc. dormitory facility. 3. Aner Dam Maharas Dhule 21.35726111 75.04883333 1986 82.94 Some There is one Deputy Wildlife htra common Irrigation Conservat Sanctuary animals Bunglow at or of found here dam site. Forests, are Barking Other 10, Deer's,Chika options Paripurti ras, Hares, include Building, Porcupines Forest Talak and Jungle Houses at Nagar, Cats. Rohini and Sawarkar Monitor Chopada Chowk, Lizard is the and a PWD Aurangaba common Bungalow at d 431005 reptile in this Shirpur. sanctuary. A large variety of birds can also be seen here. The permanent residents of the sanctuary include Egrets, Qualis, Owls, Partridges, Peafs, Herons, Corts, Spot Bills, Cormorants, Eagle Hamers and Owls etc. During the winter months, the sanctuary attracts a lot of migratory birds, primarily Cranes, Stokes, Brahming Ducks and many Waders. 4. Bhamraga Maharas Gadchir 19.53730278 80.67721111 1997 104.38 Various wild rh htra oli animals like Wildlife leopard, Sanctuary jungle fowl, wild boar and sloth bear, barking deer, blue bull, peacock and flying squirrel. -

Nagzira Nature Camp
+91-8380871850 Nagzira Nature Camp https://www.indiamart.com/nagzira-nature-camp/ Nagzira Nature Camp...A place for Tiger, Leopard, Wild dogs, Honey Badger, Sloth Bears, other Endangered Species and more than 200 different species of birds. About Us A first luxury Wildlife Camp in Nagzira - Nagzira Nature Camp. Our camp is located just 700 mtrs. from Chorkhamara Gate. Our Wildlife Camp having AC luxury tents with well appointed modern amenities. All tents are eco-friendly with beautiful surroundings of mountains and nature. Nagzira Wildlife Sanctuary is locked in the arms of nature and adorned with a picturesque landscape, luxuriant vegetation and serves as a living outdoor museum to explore and appreciate nature. The sanctuary has a number of fish, 34 species of mammals, 166 species of birds, 36 species of reptiles and four species of amphibians. The invertebrate fauna includes, besides a number of insects and ant species, several species of butterflies. Nearly 30,000 tourists visit this sanctuary annually. Wild animals to spot are the tiger, panther, bison, sambar, nilgai, chital, wild boar, sloth bear and wild dog. Presently the sanctuary extends over an area of 152.81 sq. km. (Old R.F.). this entire area is at present divided into 2 Range, 4 Rounds and 18 Beats form management point of view. This entire area is declared as a Nagzira Wildlife Sanctuary dated 3 June 1970. Camp Facilities For more information, please visit https://www.indiamart.com/nagzira-nature-camp/aboutus.html F a c t s h e e t Year of Establishment : 1970 Nature of Business : Service Provider CONTACT US Nagzira Nature Camp Contact Person: Manager Village Chorkarama, Gondia Tirora - 211011, Maharashtra, India +91-8380871850 https://www.indiamart.com/nagzira-nature-camp/. -

Wildlife Conservation Trust Update Quarter 1 | April
WILDLIFE CONSERVATION TRUST UPDATE QUARTER 1 | APRIL – JUNE 2017 Preamble The Result Based Management (RBM) is an internal monitoring mechanism that has an inbuilt system to track results and evaluate programs and projects. The RBM documents all activities within a time frame and an accountability framework and incorporates lessons learned in the annual planning process. The Annual Work Plan (AWP) for the year 2017-18 was developed as part of this RBM approach through plotting of Project Implementation Plans (PIPs); fleshing out activities in detail which lead to concrete outputs and working towards the achievement of long term outcomes for the four key verticals that fall within the overall mission and vision statements of WCT. Performance indicators were plotted as a measure to hold key personnel accountable for each and every activity within the PIP. The AWP and PIP relate the activities to the budgets and funding sources, help monitor and track the progress of the activities and eventually form the foundation of periodic reviews. These periodic reviews are key tools for capturing deviations or exceptional progress if any, and play a key role in strengthening the overall performance of the vertical. This ultimately leads to production of reports as part of the management information system framework. Conservation Research Wildlife Conservation Trust (WCT) supports forest departments by monitoring the presence and dispersal of tigers using scientifically robust techniques such as pattern matching from photographic records, radio-telemetry, and genetics among others. WCT uses camera traps to count tigers and leopards, and helps the government maintain a consolidated database of large carnivores living both inside and outside Protected Areas (PAs). -

Navegaon-Nagzira Tiger Reserve: Maharashtra
Navegaon-Nagzira Tiger Reserve: Maharashtra drishtiias.com/printpdf/navegaon-nagzira-tiger-reserve-maharashtra Why in News Three labourers were killed and two others injured during an operation to douse a forest fire at Navegaon-Nagzira Tiger Reserve (NNTR) in Maharashtra. Key Points Location: It is situated in Gondia and Bhandara districts of Maharashtra. Gondia District shares common boundaries with the state of Madhya Pradesh and Chhattisgarh in the north and eastern side respectively. Strategically, the Tiger Reserve is located in the heart of central Indian Tiger landscape which contributes almost one sixth of the total tiger population of the country. Formation: It was notified as the 46th Tiger Reserve of India in December 2013. NNTR comprised of the notified area of Nawegaon National Park, Nawegaon Wildlife Sanctuary, Nagzira Wildlife Sanctuary, New Nagzira Wildlife Sanctuary and Koka Wildlife Sanctuary. Connectivity: NNTR has connectivity with the major tiger reserves in Central India like, Kanha and Pench tiger reserve in Madhya Pradesh, Tadoba-Andhari Tiger reserve in Maharashtra, Indravati Tiger Reserve in Chhattisgarh, Indirectly with the Kawal and Nagarjuna Sagar in Telangana and Andhra Pradesh and, Achanakmar Tiger reserve in Chhattisgarh. It is also connected to important tiger bearing areas like Umred-Karhandla sanctuary and Brahampuri Division (Maharashtra). 1/3 Flora: The major forest type is "Southern Tropical Dry Deciduous Forest". Few thorny plants are also found. Bamboo occurs in abundance. Fauna: Large Carnivores such as leopards and smaller carnivores like wild dogs, wolf jackals, jungle cats and also the good population of sloth bears are seen. The important herbivore includes Cheetal, Sambar, Nilgai, Chousingha, Barking deer, Wild pig and Indian gaur. -

Standardization and Validation of a Panel of Cross-Species Microsatellites To
bioRxiv preprint doi: https://doi.org/10.1101/447201; this version posted October 18, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Standardization and validation of a panel of cross-species microsatellites to 2 individually identify the Asiatic wild dog (Cuon alpinus): implications in 3 population estimation and dynamics 4 * * *# 5 Shrushti Modi , Bilal Habib , Pallavi Ghaskadbi, Parag Nigam, Samrat Mondol 6 7 Email: 8 [email protected] 9 [email protected] 10 [email protected] 11 [email protected] 12 [email protected] 13 14 Affiliation: Wildlife Institute of India, Chandrabani, Dehradun, Uttarakhand, India 248001 15 *Equal contribution 16 17 #Corresponding author: Samrat Mondol, Animal Ecology and Conservation Biology 18 Department, Wildlife Institute of India, Chandrabani, Dehradun, Uttarakhand, India 248001 19 Email: [email protected] 20 21 22 23 24 25 Keywords: Dhole, individual identification, non-invasive sampling, genetic connectivity, 26 population parameters, Maharashtra tiger landscape 1 bioRxiv preprint doi: https://doi.org/10.1101/447201; this version posted October 18, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 27 Abstract 28 Background 29 The Asiatic wild dog or dhole (Cuon alpinus) is a highly elusive, monophyletic, forest 30 dwelling, social canid distributed across south and Southeast Asia. -

Rescue Data Mapping Project
REPORT ON “MAPPING OF WILD ANIMAL RESCUE AND ROAD KILL DATA WITHIN THE MMR USING THE PRAANI MITRA SOFTWARE APP OF THE THANE VAN VIBHAG” Submitted by: Table of Contents 1. Introduction ........................................................................................................................................ 4 2. Scope of Work and Timeline ............................................................................................................... 6 3. Literature Review ................................................................................................................................ 7 4. Study Area ......................................................................................................................................... 20 5. Methodology ..................................................................................................................................... 21 5.1: Identification of NGOs and Individual Rescuers ........................................................................ 21 5.2 Data Collection ............................................................................................................................ 21 5.3 Digitization of Data ..................................................................................................................... 21 5.4 Data Feeding into the Forest Department App .......................................................................... 21 5.5 Report preparation ....................................................................................................................