TRAF6, a Key Regulator of Tgfβ-Induced Oncogenesis in Prostate Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Megakaryopoiesis in Dengue Virus Infected K562 Cell Promotes Viral Replication Which Inhibits 2 Endomitosis and Accumulation of ROS Associated with Differentiation
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.25.172544; this version posted June 26, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Megakaryopoiesis in Dengue virus infected K562 cell promotes viral replication which inhibits 2 endomitosis and accumulation of ROS associated with differentiation 3 Jaskaran Kaur *1, Yogita Rawat *1, Vikas Sood 2, Deepak Rathore1, Shrikant K. Kumar1, Niraj K. Kumar1 4 and Sankar Bhattacharyya1 5 1 Translational Health Science and Technology Institute, NCR Biotech Science Cluster, PO Box# 4, 6 Faridabad-Gurgaon expressway, Faridabad, Haryana-121001, India 7 2 Department of Biochemistry, School of Chemical and Life Sciences, Jamia Hamdard (Hamdard 8 University) Hamdard Nagar, New Delhi - 110062, India 9 10 *Equal contribution 11 Email for correspondence: [email protected] 12 13 14 Keywords: Dengue virus replication, Megakaryopoiesis, Reactive oxygen species, Endomitosis 15 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.25.172544; this version posted June 26, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 16 Abstract: In the human host blood Monocytes and bone marrow Megakaryocytes are implicated as major 17 sites supporting high replication. The human K562 cell line supports DENV replication and represent 18 Megakaryocyte-Erythrocyte progenitors (MEP), replicating features of in vivo Megakaryopoiesis upon 19 stimulation with Phorbol esters. In this article, we report results that indicate the mutual influence of 20 Megakaryopoiesis and DENV replication on each other, through comparison of PMA-induced 21 differentiation of either mock-infected or DENV-infected K562 cells. -

Expression of the Tumor Necrosis Factor Receptor-Associated Factors
Expression of the Tumor Necrosis Factor Receptor- Associated Factors (TRAFs) 1 and 2 is a Characteristic Feature of Hodgkin and Reed-Sternberg Cells Keith F. Izban, M.D., Melek Ergin, M.D, Robert L. Martinez, B.A., HT(ASCP), Serhan Alkan, M.D. Department of Pathology, Loyola University Medical Center, Maywood, Illinois the HD cell lines. Although KMH2 showed weak Tumor necrosis factor receptor–associated factors expression, the remaining HD cell lines also lacked (TRAFs) are a recently established group of proteins TRAF5 protein. These data demonstrate that consti- involved in the intracellular signal transduction of tutive expression of TRAF1 and TRAF2 is a charac- several members of the tumor necrosis factor recep- teristic feature of HRS cells from both patient and tor (TNFR) superfamily. Recently, specific members cell line specimens. Furthermore, with the excep- of the TRAF family have been implicated in promot- tion of TRAF1 expression, HRS cells from the three ing cell survival as well as activation of the tran- HD cell lines showed similar TRAF protein expres- scription factor NF- B. We investigated the consti- sion patterns. Overall, these findings demonstrate tutive expression of TRAF1 and TRAF2 in Hodgkin the expression of several TRAF proteins in HD. Sig- and Reed–Sternberg (HRS) cells from archived nificantly, the altered regulation of selective TRAF paraffin-embedded tissues obtained from 21 pa- proteins may reflect HRS cell response to stimula- tients diagnosed with classical Hodgkin’s disease tion from the microenvironment and potentially (HD). In a selective portion of cases, examination of contribute both to apoptosis resistance and cell HRS cells for Epstein-Barr virus (EBV)–encoded maintenance of HRS cells. -

TRAF6, a Molecular Bridge Spanning Adaptive Immunity, Innate Immunity and Osteoimmunology Hao Wu1* and Joseph R
Review articles TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology Hao Wu1* and Joseph R. Arron2 Summary receptor/Toll-like receptor (IL-1R/TLR) superfamily. Gene Tumor necrosis factor (TNF) receptor associated factor targeting experiments have identified several indispen- 6 (TRAF6) is a crucial signaling molecule regulating a sable physiological functions of TRAF6, and structural diverse array of physiological processes, including and biochemical studies have revealed the potential adaptive immunity, innate immunity, bone metabolism mechanisms of its action. By virtue of its many signaling and the development of several tissues including lymph roles, TRAF6 represents an important target in the regu- nodes, mammary glands, skin and the central nervous lation of many disease processes, including immunity, system. It is a member of a group of six closely related inflammation and osteoporosis. BioEssays 25:1096– TRAF proteins, which serve as adapter molecules, coupl- 1105, 2003. ß 2003 Wiley Periodicals, Inc. ing the TNF receptor (TNFR) superfamily to intracellular signaling events. Among the TRAF proteins, TRAF6 is unique in that, in addition to mediating TNFR family Introduction signaling, it is also essential for signaling downstream of The tumor necrosis factor (TNF) receptor associated factors an unrelated family of receptors, the interleukin-1 (IL-1) (TRAFs) were first identified as two intracellular proteins, TRAF1 and TRAF2, associated with TNF-R2,(1) a member of 1Department of Biochemistry, Weill Medical College of Cornell the TNF receptor (TNFR) superfamily. There are currently six University, New York. mammalian TRAFs (TRAF1-6), which have emerged as 2Tri-Institutional MD-PhD Program, Weill Medical College of Cornell important proximal signal transducers for the TNFR super- University, New York. -

Association of Polymorphisms in the Myd88, IRAK4 and TRAF6 Genes
Molecular and Cellular Endocrinology 429 (2016) 114e119 Contents lists available at ScienceDirect Molecular and Cellular Endocrinology journal homepage: www.elsevier.com/locate/mce Association of polymorphisms in the MyD88, IRAK4 and TRAF6 genes and susceptibility to type 2 diabetes mellitus and diabetic nephropathy in a southern Han Chinese population Congcong Guo a, 1, Liju Zhang a, 1, Lihong Nie b, 1, Na Zhang a, Di Xiao a, Xingguang Ye a, Meiling Ou a, Yang Liu a, Baohuan Zhang a, Man Wang a, Hansheng Lin c, ** * Guang Yang d, e, , Chunxia Jing a, e, a Department of Epidemiology, School of Medicine, Jinan University, Guangzhou, 510632, China b Department of Endocrine, The First Affiliated Hospital of Jinan University, Guangzhou 510632, China c Department of Medical Statistics, School of Medicine, Jinan University, Guangzhou 510632, China d Department of Parasitology, School of Medicine, Jinan University, Guangzhou 510632, China e Key Laboratory of Environmental Exposure and Health in Guangzhou, Jinan University, Guangzhou 510632, China article info abstract Article history: Type 2 diabetes mellitus (T2DM) has been linked to a state of low-grade inflammation resulting from Received 17 February 2016 abnormalities in the innate immune pathway. MyD88 is an essential adaptor protein for TLR signaling, Received in revised form which is involved in activating NF-kB through IRAK4 and TRAF6. To investigate the effects of the MyD88, 5 April 2016 IRAK4 and TRAF6 polymorphisms in the susceptibility of T2DM and diabetic vascular complications, Accepted 6 April 2016 eight SNPs were analyzed in 553 T2DM patients and 553 matched healthy controls. Gene-gene in- Available online 8 April 2016 teractions and haplotype associations were also evaluated. -

DDIAS Promotes STAT3 Activation by Preventing STAT3 Recruitment To
Im et al. Oncogenesis (2020)9:1 https://doi.org/10.1038/s41389-019-0187-2 Oncogenesis ARTICLE Open Access DDIAS promotes STAT3 activation by preventing STAT3 recruitment to PTPRM in lung cancer cells Joo-Young Im 1,Bo-KyungKim 1,Kang-WooLee2, So-Young Chun1, Mi-Jung Kang1 and Misun Won1,3 Abstract DNA damage-induced apoptosis suppressor (DDIAS) regulates cancer cell survival. Here we investigated the involvement of DDIAS in IL-6–mediated signaling to understand the mechanism underlying the role of DDIAS in lung cancer malignancy. We showed that DDIAS promotes tyrosine phosphorylation of signal transducer and activator of transcription 3 (STAT3), which is constitutively activated in malignant cancers. Interestingly, siRNA protein tyrosine phosphatase (PTP) library screening revealed protein tyrosine phosphatase receptor mu (PTPRM) as a novel STAT3 PTP. PTPRM knockdown rescued the DDIAS-knockdown-mediated decrease in STAT3 Y705 phosphorylation in the presence of IL-6. However, PTPRM overexpression decreased STAT3 Y705 phosphorylation. Moreover, endogenous PTPRM interacted with endogenous STAT3 for dephosphorylation at Y705 following IL-6 treatment. As expected, PTPRM bound to wild-type STAT3 but not the STAT3 Y705F mutant. PTPRM dephosphorylated STAT3 in the absence of DDIAS, suggesting that DDIAS hampers PTPRM/STAT3 interaction. In fact, DDIAS bound to the STAT3 transactivation domain (TAD), which competes with PTPRM to recruit STAT3 for dephosphorylation. Thus we show that DDIAS prevents PTPRM/STAT3 binding and blocks STAT3 Y705 dephosphorylation, thereby sustaining STAT3 activation in lung cancer. DDIAS expression strongly correlates with STAT3 phosphorylation in human lung cancer cell lines and tissues. Thus DDIAS may be considered as a potential biomarker and therapeutic target in malignant lung cancer cells with aberrant STAT3 activation. -

Discovery of Small Molecule CD40−TRAF6 Inhibitors † ‡ § ∥ ∥ ⊥ Barbara Zarzycka, Tom Seijkens, Sander B
Article pubs.acs.org/jcim Discovery of Small Molecule CD40−TRAF6 Inhibitors † ‡ § ∥ ∥ ⊥ Barbara Zarzycka, Tom Seijkens, Sander B. Nabuurs, , Tina Ritschel, Jochen Grommes, # † ‡ † # Oliver Soehnlein, Roy Schrijver, Claudia M. van Tiel, Tilman M. Hackeng, Christian Weber, ∇ ∇ ‡ # ∥ † Fabian Giehler, Arnd Kieser, Esther Lutgens, , Gert Vriend, and Gerry A. F. Nicolaes*, † Department of Biochemistry, Cardiovascular Research Institute Maastricht (CARIM), Maastricht University, 6200 MD Maastricht, The Netherlands ‡ Department of Medical Biochemistry, Subdivision of Experimental Vascular Biology, Academic Medical Center, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands § Lead Pharma Medicine, 5349 AC Oss, The Netherlands ∥ CMBI, Radboud University Medical Centre, P.O. Box 9101, 6500 HB Nijmegen, The Netherlands ⊥ European Vascular Center Aachen-Maastricht, RWTH Aachen, 52062 Aachen, Germany # Institute for Cardiovascular Prevention, Ludwig Maximilians University, 80336 Munich, Germany ∇ Research Unit Gene Vectors, Helmholtz Zentrum München − German Research Center for Environmental Health, 81377 Munich, Germany *S Supporting Information ABSTRACT: The CD154−CD40 receptor complex plays a pivotal role in several inflammatory pathways. Attempts to inhibit the formation of this complex have resulted in systemic side effects. Downstream inhibition of the CD40 signaling pathway therefore seems a better way to ameliorate inflammatory disease. To relay a signal, the CD40 receptor recruits adapter proteins called tumor necrosis factor receptor-associated factors (TRAFs). CD40−TRAF6 interactions are known to play an essential role in several inflammatory diseases. We used in silico, in vitro, and in vivo experiments to identify and characterize compounds that block CD40−TRAF6 interactions. We present in detail our drug docking and optimization pipeline and show how we used it to find lead compounds that reduce inflammation in models of peritonitis and sepsis. -

TNF Receptor-Associated Factor 6 Is an Essential Mediator of CD40
TNF Receptor-Associated Factor 6 Is an Essential Mediator of CD40-Activated Proinflammatory Pathways in Monocytes and Macrophages This information is current as of September 26, 2021. Lata Mukundan, Gail A. Bishop, Kimberly Z. Head, Lihua Zhang, Larry M. Wahl and Jill Suttles J Immunol 2005; 174:1081-1090; ; doi: 10.4049/jimmunol.174.2.1081 http://www.jimmunol.org/content/174/2/1081 Downloaded from References This article cites 55 articles, 28 of which you can access for free at: http://www.jimmunol.org/content/174/2/1081.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 26, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology TNF Receptor-Associated Factor 6 Is an Essential Mediator of CD40-Activated Proinflammatory Pathways in Monocytes and Macrophages1 Lata Mukundan,* Gail A. Bishop,† Kimberly Z. Head,* Lihua Zhang,* Larry M. -
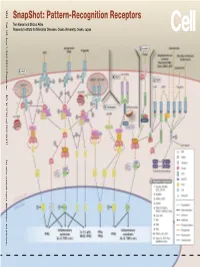
Snapshot: Pattern-Recognition Receptors
SnapShot: Pattern-Recognition Receptors SnapShot:Pattern-Recognition Kawai and Shizuo Akira Taro Osaka, Japan Diseases, Osaka University, Institute for Microbial Research 1024 Cell 129, June 1, 2007 ©2007 Elsevier Inc. DOI 10.1016/j.cell.2007.05.017 See online version for legend, abbreviations, and references. SnapShot: Pattern-Recognition Receptors Taro Kawai and Shizuo Akira Research Institute for Microbial Diseases, Osaka University, Osaka, Japan (A) Toll-like receptor signaling. Toll-like receptor (TLR) 3 recognizes polyinosinic-polycytidylic acid (poly IC), whereas TLR4 recognizes lipopolysaccharide (LPS). TLR2 recog- nizes various components such as lipoprotein and peptidoglycan (PGN). TLR5 detects flagellin. TLR7 and TLR9 detect single-stranded (ss)RNA and CpG DNA, respectively. Each TLR recruits a distinct set of Toll/interleukin-1 receptor (TIR) domain-containing adaptor molecules such as myeloid differentiation primary response gene 88 (MyD88), TIR-containing adaptor protein (TIRAP, also known as MAL), TIR-containing adaptor-inducing IFNβ (TRIF, also known as TICAM1) and TRIF-related adaptor molecule (TRAM, also known as TICAM2). TLR3 uses TRIF, and TLR5, 7, and 9 use MyD88. TLR2 uses MyD88 and TIRAP, and TLR4 uses MyD88, TIRAP, TRIF, and TRAM. MyD88 binds to inter- leukin-1 receptor-associated kinase 4 (IRAK4) and TRAF6. TRIF binds receptor-interacting protein 1 (RIP1) and TRAF6. TRAF6 forms a complex with Ubc13, Uev1A, and ECSIT (evolutionarily conserved signaling intermediate in Toll/IL-1R pathways) to activate a complex containing transforming growth factor-β-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), TAB2, and TAB3. TAK1 activates IκB kinase (IKK) complex consisting of IKKα, IKKβ, and Nemo (also known as IKKγ), which results in the phosphorylation and proteasomal degradation of IκB proteins and the release of a transcription factor NFκB to the nucleus to regulate expression of inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor α (TNFα). -

THE ROLE of TRAF6 PHOSPHORYLATION in Src/ TRAF6-MEDIATED IKK, JNK, Akt ACTIVATION and TUMORIGENESIS
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 8-2014 THE ROLE OF TRAF6 PHOSPHORYLATION IN Src/ TRAF6-MEDIATED IKK, JNK, Akt ACTIVATION AND TUMORIGENESIS Yun Seong Jeong Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Biological Phenomena, Cell Phenomena, and Immunity Commons, and the Biology Commons Recommended Citation Jeong, Yun Seong, "THE ROLE OF TRAF6 PHOSPHORYLATION IN Src/TRAF6-MEDIATED IKK, JNK, Akt ACTIVATION AND TUMORIGENESIS" (2014). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 494. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/494 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. THE ROLE OF TRAF6 PHOSPHORYLATION IN Src/TRAF6-MEDIATED IKK, JNK, Akt ACTIVATION AND TUMORIGENESIS by Yun Seong -

TRAF6 Phosphorylation Prevents Its Autophagic Degradation and Re-Shapes LPS-Triggered Signaling Networks
cancers Article TRAF6 Phosphorylation Prevents Its Autophagic Degradation and Re-Shapes LPS-Triggered Signaling Networks Julia Busch 1, Rita Moreno 2, Laureano de la Vega 2 , Vera Vivian Saul 1, Susanne Bacher 1, Felix von Zweydorf 3 , Marius Ueffing 4 , Axel Weber 5 , Christian Johannes Gloeckner 3,6 , Uwe Linne 7 , Michael Kracht 5 and Michael Lienhard Schmitz 1,* 1 Institute of Biochemistry, Member of the German Center for Lung Research, Justus Liebig University, 35392 Giessen, Germany; [email protected] (J.B.); [email protected] (V.V.S.); [email protected] (S.B.) 2 Division of Cellular Medicine, Ninewells Hospital and Medical School, University of Dundee, James Arrott Drive, Dundee DD1 9SY, UK; [email protected] (R.M.); [email protected] (L.d.l.V.) 3 German Center for Neurodegenerative Diseases (DZNE), 72076 Tübingen, Germany; [email protected] (F.v.Z.); [email protected] (C.J.G.) 4 Centre for Ophthalmology, Institute for Ophthalmic Research, University of Tübingen, 72076 Tübingen, Germany; marius.ueffi[email protected] 5 Rudolf Buchheim Institute of Pharmacology, Member of the German Center for Lung Research, Justus Liebig University, 35392 Giessen, Germany; [email protected] (A.W.); [email protected] (M.K.) 6 Core Facility for Medical Bioanalytics, Center for Ophthalmology, Institute for Ophthalmic Research, University of Tübingen, 72076 Tübingen, Germany 7 Mass Spectrometry Facility of the Department of Chemistry, Philipps University, 35043 Marburg, Germany; [email protected] Citation: Busch, J.; Moreno, R.; de la * Correspondence: [email protected] Vega, L.; Saul, V.V.; Bacher, S.; von Zweydorf, F.; Ueffing, M.; Weber, A.; Simple Summary: Here, we reveal that basal turnover and autophagy-induced decay of the ubiquitin Gloeckner, C.J.; Linne, U.; et al. -

The Role of Janus Kinase 3 in CD4+ T Cell Homeostasis and Function: a Dissertation
University of Massachusetts Medical School eScholarship@UMMS GSBS Dissertations and Theses Graduate School of Biomedical Sciences 2004-09-13 The role of Janus Kinase 3 in CD4+ T Cell Homeostasis and Function: A Dissertation Shane Renee Mayack University of Massachusetts Medical School Let us know how access to this document benefits ou.y Follow this and additional works at: https://escholarship.umassmed.edu/gsbs_diss Part of the Amino Acids, Peptides, and Proteins Commons, Cells Commons, Enzymes and Coenzymes Commons, and the Hemic and Immune Systems Commons Repository Citation Mayack SR. (2004). The role of Janus Kinase 3 in CD4+ T Cell Homeostasis and Function: A Dissertation. GSBS Dissertations and Theses. https://doi.org/10.13028/4qq7-k514. Retrieved from https://escholarship.umassmed.edu/gsbs_diss/94 This material is brought to you by eScholarship@UMMS. It has been accepted for inclusion in GSBS Dissertations and Theses by an authorized administrator of eScholarship@UMMS. For more information, please contact [email protected]. THE ROLE OF JANUS KINASE 3 IN CD4+ T CELL HOMEOSTASIS AND FUNCTION A Dissertation Presented BY Shane Renee Mayack Submitted to the Faculty of the University of Massachusetts Graduate School of Biomedical Sciences, Worcester in partial llfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY September 13,2004 Interdisciplinary Graduate Program Department of Pathology Program in ImmunologyiVirology Copyright Information The content of this dissertation has appeared in the following publication: Mayack, S. Berg., L. (2004) Alternative CD4+ T cell differentiation in the absence of Jak3. Submitted for publication. Joonsoo Kang, Chair of Committee Alan Rothman, Member of Committee Robert Woodland, Member of Committee Katherine Luzuriaga, Member of Committee Gordon Freeman, Member of Committee Leslie J. -

The Ubiquitin E3 Ligase TRAF6 Exacerbates Pathological Cardiac Hypertrophy Via TAK1-Dependent Signalling
ARTICLE Received 9 Sep 2015 | Accepted 7 Mar 2016 | Published 1 Jun 2016 DOI: 10.1038/ncomms11267 OPEN The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1-dependent signalling Yan-Xiao Ji1,2,3,*, Peng Zhang1,2,3,*, Xiao-Jing Zhang1,2,3,*, Yi-Chao Zhao4, Ke-Qiong Deng1,2,3, Xi Jiang1,2,3, Pi-Xiao Wang1,2,3, Zan Huang5 & Hongliang Li1,2,3 Tumour necrosis factor receptor-associated factor 6 (TRAF6) is a ubiquitin E3 ligase that regulates important biological processes. However, the role of TRAF6 in cardiac hypertrophy remains unknown. Here, we show that TRAF6 levels are increased in human and murine hypertrophied hearts, which is regulated by reactive oxygen species (ROS) production. Cardiac-specific Traf6 overexpression exacerbates cardiac hypertrophy in response to pressure overload or angiotensin II (Ang II) challenge, whereas Traf6 deficiency causes an alleviated hypertrophic phenotype in mice. Mechanistically, we show that ROS, generated during hypertrophic progression, triggers TRAF6 auto-ubiquitination that facilitates recruitment of TAB2 and its binding to transforming growth factor beta-activated kinase 1 (TAK1), which, in turn, enables the direct TRAF6–TAK1 interaction and promotes TAK1 ubiquitination. The binding of TRAF6 to TAK1 and the induction of TAK1 ubiquitination and activation are indispensable for TRAF6-regulated cardiac remodelling. Taken together, we define TRAF6 as an essential molecular switch leading to cardiac hypertrophy in a TAK1-dependent manner. 1 Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan 430060, China. 2 Animal Experiment Center/Animal Biosafety Level-III Laboratory, Wuhan University, Wuhan 430060, China. 3 Medical Research Institute, School of Medicine, Wuhan University, Wuhan 430071, China.