The Python-Based Simulations of Chemistry Framework (Pyscf)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Free and Open Source Software for Computational Chemistry Education
Free and Open Source Software for Computational Chemistry Education Susi Lehtola∗,y and Antti J. Karttunenz yMolecular Sciences Software Institute, Blacksburg, Virginia 24061, United States zDepartment of Chemistry and Materials Science, Aalto University, Espoo, Finland E-mail: [email protected].fi Abstract Long in the making, computational chemistry for the masses [J. Chem. Educ. 1996, 73, 104] is finally here. We point out the existence of a variety of free and open source software (FOSS) packages for computational chemistry that offer a wide range of functionality all the way from approximate semiempirical calculations with tight- binding density functional theory to sophisticated ab initio wave function methods such as coupled-cluster theory, both for molecular and for solid-state systems. By their very definition, FOSS packages allow usage for whatever purpose by anyone, meaning they can also be used in industrial applications without limitation. Also, FOSS software has no limitations to redistribution in source or binary form, allowing their easy distribution and installation by third parties. Many FOSS scientific software packages are available as part of popular Linux distributions, and other package managers such as pip and conda. Combined with the remarkable increase in the power of personal devices—which rival that of the fastest supercomputers in the world of the 1990s—a decentralized model for teaching computational chemistry is now possible, enabling students to perform reasonable modeling on their own computing devices, in the bring your own device 1 (BYOD) scheme. In addition to the programs’ use for various applications, open access to the programs’ source code also enables comprehensive teaching strategies, as actual algorithms’ implementations can be used in teaching. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

Open Babel Documentation Release 2.3.1
Open Babel Documentation Release 2.3.1 Geoffrey R Hutchison Chris Morley Craig James Chris Swain Hans De Winter Tim Vandermeersch Noel M O’Boyle (Ed.) December 05, 2011 Contents 1 Introduction 3 1.1 Goals of the Open Babel project ..................................... 3 1.2 Frequently Asked Questions ....................................... 4 1.3 Thanks .................................................. 7 2 Install Open Babel 9 2.1 Install a binary package ......................................... 9 2.2 Compiling Open Babel .......................................... 9 3 obabel and babel - Convert, Filter and Manipulate Chemical Data 17 3.1 Synopsis ................................................. 17 3.2 Options .................................................. 17 3.3 Examples ................................................. 19 3.4 Differences between babel and obabel .................................. 21 3.5 Format Options .............................................. 22 3.6 Append property values to the title .................................... 22 3.7 Filtering molecules from a multimolecule file .............................. 22 3.8 Substructure and similarity searching .................................. 25 3.9 Sorting molecules ............................................ 25 3.10 Remove duplicate molecules ....................................... 25 3.11 Aliases for chemical groups ....................................... 26 4 The Open Babel GUI 29 4.1 Basic operation .............................................. 29 4.2 Options ................................................. -

The Alexandria Library, a Quantum-Chemical Database of Molecular Properties for Force field Development 9 2017 Received: October 1 1 1 Mohammad M
www.nature.com/scientificdata OPEN Data Descriptor: The Alexandria library, a quantum-chemical database of molecular properties for force field development 9 2017 Received: October 1 1 1 Mohammad M. Ghahremanpour , Paul J. van Maaren & David van der Spoel Accepted: 19 February 2018 Published: 10 April 2018 Data quality as well as library size are crucial issues for force field development. In order to predict molecular properties in a large chemical space, the foundation to build force fields on needs to encompass a large variety of chemical compounds. The tabulated molecular physicochemical properties also need to be accurate. Due to the limited transparency in data used for development of existing force fields it is hard to establish data quality and reusability is low. This paper presents the Alexandria library as an open and freely accessible database of optimized molecular geometries, frequencies, electrostatic moments up to the hexadecupole, electrostatic potential, polarizabilities, and thermochemistry, obtained from quantum chemistry calculations for 2704 compounds. Values are tabulated and where available compared to experimental data. This library can assist systematic development and training of empirical force fields for a broad range of molecules. Design Type(s) data integration objective • molecular physical property analysis objective Measurement Type(s) physicochemical characterization Technology Type(s) Computational Chemistry Factor Type(s) Sample Characteristic(s) 1 Uppsala Centre for Computational Chemistry, Science for Life Laboratory, Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, SE-75124 Uppsala, Sweden. Correspondence and requests for materials should be addressed to D.v.d.S. (email: [email protected]). -
![Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021](https://docslib.b-cdn.net/cover/7378/automated-construction-of-quantum-classical-hybrid-models-arxiv-2102-09355v1-physics-chem-ph-18-feb-2021-177378.webp)
Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021
Automated construction of quantum{classical hybrid models Christoph Brunken and Markus Reiher∗ ETH Z¨urich, Laboratorium f¨urPhysikalische Chemie, Vladimir-Prelog-Weg 2, 8093 Z¨urich, Switzerland February 18, 2021 Abstract We present a protocol for the fully automated construction of quantum mechanical-(QM){ classical hybrid models by extending our previously reported approach on self-parametri- zing system-focused atomistic models (SFAM) [J. Chem. Theory Comput. 2020, 16 (3), 1646{1665]. In this QM/SFAM approach, the size and composition of the QM region is evaluated in an automated manner based on first principles so that the hybrid model describes the atomic forces in the center of the QM region accurately. This entails the au- tomated construction and evaluation of differently sized QM regions with a bearable com- putational overhead that needs to be paid for automated validation procedures. Applying SFAM for the classical part of the model eliminates any dependence on pre-existing pa- rameters due to its system-focused quantum mechanically derived parametrization. Hence, QM/SFAM is capable of delivering a high fidelity and complete automation. Furthermore, since SFAM parameters are generated for the whole system, our ansatz allows for a con- venient re-definition of the QM region during a molecular exploration. For this purpose, a local re-parametrization scheme is introduced, which efficiently generates additional clas- sical parameters on the fly when new covalent bonds are formed (or broken) and moved to the classical region. arXiv:2102.09355v1 [physics.chem-ph] 18 Feb 2021 ∗Corresponding author; e-mail: [email protected] 1 1 Introduction In contrast to most protocols of computational quantum chemistry that consider isolated molecules, chemical processes can take place in a vast variety of complex environments. -

Popular GPU-Accelerated Applications
LIFE & MATERIALS SCIENCES GPU-ACCELERATED APPLICATIONS | CATALOG | AUG 12 LIFE & MATERIALS SCIENCES APPLICATIONS CATALOG Application Description Supported Features Expected Multi-GPU Release Status Speed Up* Support Bioinformatics BarraCUDA Sequence mapping software Alignment of short sequencing 6-10x Yes Available now reads Version 0.6.2 CUDASW++ Open source software for Smith-Waterman Parallel search of Smith- 10-50x Yes Available now protein database searches on GPUs Waterman database Version 2.0.8 CUSHAW Parallelized short read aligner Parallel, accurate long read 10x Yes Available now aligner - gapped alignments to Version 1.0.40 large genomes CATALOG GPU-BLAST Local search with fast k-tuple heuristic Protein alignment according to 3-4x Single Only Available now blastp, multi cpu threads Version 2.2.26 GPU-HMMER Parallelized local and global search with Parallel local and global search 60-100x Yes Available now profile Hidden Markov models of Hidden Markov Models Version 2.3.2 mCUDA-MEME Ultrafast scalable motif discovery algorithm Scalable motif discovery 4-10x Yes Available now based on MEME algorithm based on MEME Version 3.0.12 MUMmerGPU A high-throughput DNA sequence alignment Aligns multiple query sequences 3-10x Yes Available now LIFE & MATERIALS& LIFE SCIENCES APPLICATIONS program against reference sequence in Version 2 parallel SeqNFind A GPU Accelerated Sequence Analysis Toolset HW & SW for reference 400x Yes Available now assembly, blast, SW, HMM, de novo assembly UGENE Opensource Smith-Waterman for SSE/CUDA, Fast short -

User Manual for the Uppsala Quantum Chemistry Package UQUANTCHEM V.35
User manual for the Uppsala Quantum Chemistry package UQUANTCHEM V.35 by Petros Souvatzis Uppsala 2016 Contents 1 Introduction 6 2 Compiling the code 7 3 What Can be done with UQUANTCHEM 9 3.1 Hartree Fock Calculations . 9 3.2 Configurational Interaction Calculations . 9 3.3 M¨ollerPlesset Calculations (MP2) . 9 3.4 Density Functional Theory Calculations (DFT)) . 9 3.5 Time Dependent Density Functional Theory Calculations (TDDFT)) . 10 3.6 Quantum Montecarlo Calculations . 10 3.7 Born Oppenheimer Molecular Dynamics . 10 4 Setting up a UQANTCHEM calculation 12 4.1 The input files . 12 4.1.1 The INPUTFILE-file . 12 4.1.2 The BASISFILE-file . 13 4.1.3 The BASISFILEAUX-file . 14 4.1.4 The DENSMATSTARTGUESS.dat-file . 14 4.1.5 The MOLDYNRESTART.dat-file . 14 4.1.6 The INITVELO.dat-file . 15 4.1.7 Running Uquantchem . 15 4.2 Input parameters . 15 4.2.1 CORRLEVEL ................................. 15 4.2.2 ADEF ..................................... 15 4.2.3 DOTDFT ................................... 16 4.2.4 NSCCORR ................................... 16 4.2.5 SCERR .................................... 16 4.2.6 MIXTDDFT .................................. 16 4.2.7 EPROFILE .................................. 16 4.2.8 DOABSSPECTRUM ............................... 17 4.2.9 NEPERIOD .................................. 17 4.2.10 EFIELDMAX ................................. 17 4.2.11 EDIR ..................................... 17 4.2.12 FIELDDIR .................................. 18 4.2.13 OMEGA .................................... 18 2 CONTENTS 3 4.2.14 NCHEBGAUSS ................................. 18 4.2.15 RIAPPROX .................................. 18 4.2.16 LIMPRECALC (Only openmp-version) . 19 4.2.17 DIAGDG ................................... 19 4.2.18 NLEBEDEV .................................. 19 4.2.19 MOLDYN ................................... 19 4.2.20 DAMPING ................................... 19 4.2.21 XLBOMD .................................. -

Python Tools in Computational Chemistry (And Biology)
Python Tools in Computational Chemistry (and Biology) Andrew Dalke Dalke Scientific, AB Göteborg, Sweden EuroSciPy, 26-27 July, 2008 “Why does ‘import numpy’ take 0.4 seconds? Does it need to import 228 libraries?” - My first Numpy-discussion post (paraphrased) Your use case isn't so typical and so suffers on the import time end of the balance. - Response from Robert Kern (Others did complain. Import time down to 0.28s.) 52,000 structures PDB doubles every 2½ years HEADER PHOTORECEPTOR 23-MAY-90 1BRD 1BRD 2 COMPND BACTERIORHODOPSIN 1BRD 3 SOURCE (HALOBACTERIUM $HALOBIUM) 1BRD 4 EXPDTA ELECTRON DIFFRACTION 1BRD 5 AUTHOR R.HENDERSON,J.M.BALDWIN,T.A.CESKA,F.ZEMLIN,E.BECKMANN, 1BRD 6 AUTHOR 2 K.H.DOWNING 1BRD 7 REVDAT 3 15-JAN-93 1BRDB 1 SEQRES 1BRDB 1 REVDAT 2 15-JUL-91 1BRDA 1 REMARK 1BRDA 1 .. ATOM 54 N PRO 8 20.397 -15.569 -13.739 1.00 20.00 1BRD 136 ATOM 55 CA PRO 8 21.592 -15.444 -12.900 1.00 20.00 1BRD 137 ATOM 56 C PRO 8 21.359 -15.206 -11.424 1.00 20.00 1BRD 138 ATOM 57 O PRO 8 21.904 -15.930 -10.563 1.00 20.00 1BRD 139 ATOM 58 CB PRO 8 22.367 -14.319 -13.591 1.00 20.00 1BRD 140 ATOM 59 CG PRO 8 22.089 -14.564 -15.053 1.00 20.00 1BRD 141 ATOM 60 CD PRO 8 20.647 -15.054 -15.103 1.00 20.00 1BRD 142 ATOM 61 N GLU 9 20.562 -14.211 -11.095 1.00 20.00 1BRD 143 ATOM 62 CA GLU 9 20.192 -13.808 -9.737 1.00 20.00 1BRD 144 ATOM 63 C GLU 9 19.567 -14.935 -8.932 1.00 20.00 1BRD 145 ATOM 64 O GLU 9 19.815 -15.104 -7.724 1.00 20.00 1BRD 146 ATOM 65 CB GLU 9 19.248 -12.591 -9.820 1.00 99.00 1 1BRD 147 ATOM 66 CG GLU 9 19.902 -11.351 -10.387 1.00 99.00 1 1BRD 148 ATOM 67 CD GLU 9 19.243 -10.169 -10.980 1.00 99.00 1 1BRD 149 ATOM 68 OE1 GLU 9 18.323 -10.191 -11.782 1.00 99.00 1 1BRD 150 ATOM 69 OE2 GLU 9 19.760 -9.089 -10.597 1.00 99.00 1 1BRD 151 ATOM 70 N TRP 10 18.764 -15.737 -9.597 1.00 20.00 1BRD 152 ATOM 71 CA TRP 10 18.034 -16.884 -9.090 1.00 20.00 1BRD 153 ATOM 72 C TRP 10 18.843 -17.908 -8.318 1.00 20.00 1BRD 154 ATOM 73 O TRP 10 18.376 -18.310 -7.230 1.00 20.00 1BRD 155 . -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -
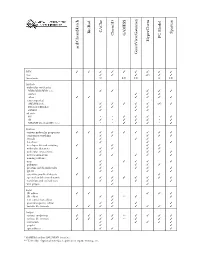
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -
![Modern Quantum Chemistry with [Open]Molcas](https://docslib.b-cdn.net/cover/4742/modern-quantum-chemistry-with-open-molcas-744742.webp)
Modern Quantum Chemistry with [Open]Molcas
Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 Submitted: 17 February 2020 . Accepted: 11 May 2020 . Published Online: 05 June 2020 Francesco Aquilante , Jochen Autschbach , Alberto Baiardi , Stefano Battaglia , Veniamin A. Borin , Liviu F. Chibotaru , Irene Conti , Luca De Vico , Mickaël Delcey , Ignacio Fdez. Galván , Nicolas Ferré , Leon Freitag , Marco Garavelli , Xuejun Gong , Stefan Knecht , Ernst D. Larsson , Roland Lindh , Marcus Lundberg , Per Åke Malmqvist , Artur Nenov , Jesper Norell , Michael Odelius , Massimo Olivucci , Thomas B. Pedersen , Laura Pedraza-González , Quan M. Phung , Kristine Pierloot , Markus Reiher , Igor Schapiro , Javier Segarra-Martí , Francesco Segatta , Luis Seijo , Saumik Sen , Dumitru-Claudiu Sergentu , Christopher J. Stein , Liviu Ungur , Morgane Vacher , Alessio Valentini , and Valera Veryazov J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 152, 214117 © 2020 Author(s). The Journal ARTICLE of Chemical Physics scitation.org/journal/jcp Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); doi: 10.1063/5.0004835 Submitted: 17 February 2020 • Accepted: 11 May 2020 • Published Online: 5 June 2020 Francesco Aquilante,1,a) Jochen Autschbach,2,b) Alberto Baiardi,3,c) Stefano Battaglia,4,d) Veniamin A. Borin,5,e) Liviu F. Chibotaru,6,f) Irene Conti,7,g) Luca De Vico,8,h) Mickaël Delcey,9,i) Ignacio Fdez. Galván,4,j) Nicolas Ferré,10,k) Leon Freitag,3,l) Marco Garavelli,7,m) Xuejun Gong,11,n) Stefan Knecht,3,o) Ernst D. Larsson,12,p) Roland Lindh,4,q) Marcus Lundberg,9,r) Per Åke Malmqvist,12,s) Artur Nenov,7,t) Jesper Norell,13,u) Michael Odelius,13,v) Massimo Olivucci,8,14,w) Thomas B. -

Benchmarking and Application of Density Functional Methods In
BENCHMARKING AND APPLICATION OF DENSITY FUNCTIONAL METHODS IN COMPUTATIONAL CHEMISTRY by BRIAN N. PAPAS (Under Direction the of Henry F. Schaefer III) ABSTRACT Density Functional methods were applied to systems of chemical interest. First, the effects of integration grid quadrature choice upon energy precision were documented. This was done through application of DFT theory as implemented in five standard computational chemistry programs to a subset of the G2/97 test set of molecules. Subsequently, the neutral hydrogen-loss radicals of naphthalene, anthracene, tetracene, and pentacene and their anions where characterized using five standard DFT treatments. The global and local electron affinities were computed for the twelve radicals. The results for the 1- naphthalenyl and 2-naphthalenyl radicals were compared to experiment, and it was found that B3LYP appears to be the most reliable functional for this type of system. For the larger systems the predicted site specific adiabatic electron affinities of the radicals are 1.51 eV (1-anthracenyl), 1.46 eV (2-anthracenyl), 1.68 eV (9-anthracenyl); 1.61 eV (1-tetracenyl), 1.56 eV (2-tetracenyl), 1.82 eV (12-tetracenyl); 1.93 eV (14-pentacenyl), 2.01 eV (13-pentacenyl), 1.68 eV (1-pentacenyl), and 1.63 eV (2-pentacenyl). The global minimum for each radical does not have the same hydrogen removed as the global minimum for the analogous anion. With this in mind, the global (or most preferred site) adiabatic electron affinities are 1.37 eV (naphthalenyl), 1.64 eV (anthracenyl), 1.81 eV (tetracenyl), and 1.97 eV (pentacenyl). In later work, ten (scandium through zinc) homonuclear transition metal trimers were studied using one DFT 2 functional.