A Combined Algorithm for Genome- Wide Prediction of Protein Function
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation
From THE DEPARTMENT OF MICROBIOLOGY TUMOR AND CELL BIOLOGY (MTC) Karolinska Institutet, Stockholm, Sweden ROLE OF MITOCHONDRIAL RIBOSOMAL PROTEIN S18-2 IN CANCEROGENESIS AND IN REGULATION OF STEMNESS AND DIFFERENTIATION Muhammad Mushtaq Stockholm 2017 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by E-Print AB 2017 © Muhammad Mushtaq, 2017 ISBN 978-91-7676-697-2 Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation THESIS FOR DOCTORAL DEGREE (Ph.D.) By Muhammad Mushtaq Principal Supervisor: Faculty Opponent: Associate Professor Elena Kashuba Professor Pramod Kumar Srivastava Karolinska Institutet University of Connecticut Department of Microbiology Tumor and Cell Center for Immunotherapy of Cancer and Biology (MTC) Infectious Diseases Co-supervisor(s): Examination Board: Professor Sonia Lain Professor Ola Söderberg Karolinska Institutet Uppsala University Department of Microbiology Tumor and Cell Department of Immunology, Genetics and Biology (MTC) Pathology (IGP) Professor George Klein Professor Boris Zhivotovsky Karolinska Institutet Karolinska Institutet Department of Microbiology Tumor and Cell Institute of Environmental Medicine (IMM) Biology (MTC) Professor Lars-Gunnar Larsson Karolinska Institutet Department of Microbiology Tumor and Cell Biology (MTC) Dedicated to my parents ABSTRACT Mitochondria carry their own ribosomes (mitoribosomes) for the translation of mRNA encoded by mitochondrial DNA. The architecture of mitoribosomes is mainly composed of mitochondrial ribosomal proteins (MRPs), which are encoded by nuclear genomic DNA. Emerging experimental evidences reveal that several MRPs are multifunctional and they exhibit important extra-mitochondrial functions, such as involvement in apoptosis, protein biosynthesis and signal transduction. Dysregulations of the MRPs are associated with severe pathological conditions, including cancer. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Candidate Regulators of Dyslipidemia in Chromosome 1
University of Groningen Candidate Regulators of Dyslipidemia in Chromosome 1 Substitution Lines Using Liver Co- Expression Profiling Analysis Xu, Fuyi; Wang, Maochun; Hu, Shixian; Zhou, Yuxun; Collyer, John; Li, Kai; Xu, Hongyan; Xiao, Junhua Published in: Frontiers in Genetics DOI: 10.3389/fgene.2019.01258 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2020 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Xu, F., Wang, M., Hu, S., Zhou, Y., Collyer, J., Li, K., Xu, H., & Xiao, J. (2020). Candidate Regulators of Dyslipidemia in Chromosome 1 Substitution Lines Using Liver Co-Expression Profiling Analysis. Frontiers in Genetics, 10, [1258]. https://doi.org/10.3389/fgene.2019.01258 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover -

Open FINAL GRAD SCHOOL.Pdf
The Pennsylvania State University The Graduate School Eberly College of Science PHOSPHOPROTEOMIC ANALYSIS OF RIBOSOMAL PROTEINS: IMPLICATIONS IN TRANSLATION AND APOPTOSIS A Dissertation in Biochemistry, Microbiology, and Molecular Biology by Jennifer Lynn Miller © 2009 Jennifer Lynn Miller Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2009 The dissertation of Jennifer Lynn Miller was reviewed and approved* by the following: Emine C. Koc Assistant Professor Biochemistry and Molecular Biology Dissertation Advisor Chair of Committee Robert A. Schlegel Professor of Biochemistry and Molecular Biology Wendy Hanna-Rose Assistant Professor Biochemistry and Molecular Biology Ming Tien Professor of Biochemistry Erin D. Sheets Assistant Professor of Chemistry Richard J. Frisque Professor of Molecular Virology Head of the Department of Biochemistry and Molecular Biology *Signatures are on file in the Graduate School. ABSTRACT Mammalian mitochondrial ribosomes synthesize thirteen proteins that are essential for oxidative phosphorylation. Besides having a major role in ATP synthesis, mitochondria also contribute to biochemical processes coordinating apoptosis, mitochondrial diseases, and aging in eukaryotic cells. This unique class of ribosomes is protein-rich and distinct from cytoplasmic ribosomes. However, mitochondrial ribosomes (55S) share a significant homology to bacterial ribosomes (70S), particularly in size, the general mechanism of translation, and ribosomal protein content. Due to the overall resemblance between the two systems and the earlier reports of post-translational modifications, we investigated how phosphorylation of ribosomal proteins from bacteria and mitochondria regulates translation and other acquired roles. Identification of twenty- four phosphorylated 70S and 55S ribosomal proteins as well as the potential endogenous kinase was achieved using 2D-gel electrophoresis and tandem mass spectrometry. -

Influenza-Specific Effector Memory B Cells Predict Long-Lived Antibody Responses to Vaccination in Humans
bioRxiv preprint doi: https://doi.org/10.1101/643973; this version posted February 18, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Influenza-specific effector memory B cells predict long-lived antibody responses to vaccination in humans Anoma Nellore1, Esther Zumaquero2, Christopher D. Scharer3, Rodney G. King2, Christopher M. Tipton4, Christopher F. Fucile5, Tian Mi3, Betty Mousseau2, John E. Bradley6, Fen Zhou2, Paul A. Goepfert1, Jeremy M. Boss3, Troy D. Randall6, Ignacio Sanz4, Alexander F. Rosenberg2,5, Frances E. Lund2 1Dept. of Medicine, Division of Infectious Disease, 2Dept of Microbiology, 5Informatics Institute, 6Dept. of Medicine, Division of Clinical Immunology and Rheumatology and at The University of Alabama at Birmingham, Birmingham, AL 35294 USA 3Dept. of Microbiology and Immunology and 4Department of Medicine, Division of Rheumatology Emory University, Atlanta, GA 30322, USA Correspondence should be addressed to: Frances E. Lund, PhD Charles H. McCauley Professor and Chair Dept of Microbiology University of Alabama at Birmingham 276 BBRB Box 11 1720 2nd Avenue South Birmingham AL 35294-2170 [email protected] SHORT RUNNING TITLE: Effector memory B cell development after influenza vaccination 1 bioRxiv preprint doi: https://doi.org/10.1101/643973; this version posted February 18, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Seasonal influenza vaccination elicits hemagglutinin (HA)-specific CD27+ memory B cells (Bmem) that differ in expression of T-bet, BACH2 and TCF7. -

Transcriptomic and Proteomic Landscape of Mitochondrial
TOOLS AND RESOURCES Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals Inge Ku¨ hl1,2†*, Maria Miranda1†, Ilian Atanassov3, Irina Kuznetsova4,5, Yvonne Hinze3, Arnaud Mourier6, Aleksandra Filipovska4,5, Nils-Go¨ ran Larsson1,7* 1Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne, Germany; 2Department of Cell Biology, Institute of Integrative Biology of the Cell (I2BC) UMR9198, CEA, CNRS, Univ. Paris-Sud, Universite´ Paris-Saclay, Gif- sur-Yvette, France; 3Proteomics Core Facility, Max Planck Institute for Biology of Ageing, Cologne, Germany; 4Harry Perkins Institute of Medical Research, The University of Western Australia, Nedlands, Australia; 5School of Molecular Sciences, The University of Western Australia, Crawley, Australia; 6The Centre National de la Recherche Scientifique, Institut de Biochimie et Ge´ne´tique Cellulaires, Universite´ de Bordeaux, Bordeaux, France; 7Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden Abstract Dysfunction of the oxidative phosphorylation (OXPHOS) system is a major cause of human disease and the cellular consequences are highly complex. Here, we present comparative *For correspondence: analyses of mitochondrial proteomes, cellular transcriptomes and targeted metabolomics of five [email protected] knockout mouse strains deficient in essential factors required for mitochondrial DNA gene (IKu¨ ); expression, leading to OXPHOS dysfunction. Moreover, -

Receptor Signaling Through Osteoclast-Associated Monocyte
Downloaded from http://www.jimmunol.org/ by guest on September 29, 2021 is online at: average * The Journal of Immunology The Journal of Immunology , 20 of which you can access for free at: 2015; 194:3169-3179; Prepublished online 27 from submission to initial decision 4 weeks from acceptance to publication February 2015; doi: 10.4049/jimmunol.1402800 http://www.jimmunol.org/content/194/7/3169 Collagen Induces Maturation of Human Monocyte-Derived Dendritic Cells by Signaling through Osteoclast-Associated Receptor Heidi S. Schultz, Louise M. Nitze, Louise H. Zeuthen, Pernille Keller, Albrecht Gruhler, Jesper Pass, Jianhe Chen, Li Guo, Andrew J. Fleetwood, John A. Hamilton, Martin W. Berchtold and Svetlana Panina J Immunol cites 43 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Author Choice option Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Freely available online through http://www.jimmunol.org/content/suppl/2015/02/27/jimmunol.140280 0.DCSupplemental This article http://www.jimmunol.org/content/194/7/3169.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Author Choice Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. -

Supplementary Figure 1 Standardization of Gene Expression
Supplementary Figure 1 Standardization of gene expression Notes: (A) Standardization of GSE86544, (B) standardization of GSE103479, (C) standardization of GSE102238, (D) Standardization of GSE7055. The blue bar represents the data before normalization, and the red bar represents the data after normalization. Supplementary Figure 2 Correlation between module eigengenes and clinical traits especially PNI in GSE103479 and GSE102238 datasets. Notes: (A, B) Module-trait relationships in GSE103479 and GSE102238 datasets. The correlation coefficients and corresponding P-values in the brackets are contained in each cell. The table is color- coded by correlation between eigengenes and traits according to the color legend on the right side. The modules with the most significant differences are displayed in brackets. Abbreviations: PNI, perineural invasion. Supplementary Figure 3 The expression values of CCNB2 in pancreatic cancer (GSE102238) and colon cancer (GSE103479). Notes: (A, B) CCNB2 expression values were detected in GSE102238 and GSE103479. Abbreviations: CCNB2, cyclin B2 Supplementary Table 1 Results of top 20 pathway enrichment analysis of GSE7055 Term Category Description Count Log10(P) Genes GO:0000280 GO Biological nuclear division 33 -23.4 BIRC5,BUB1B,CCNB1,CCNE1,CDC20, Processes CKS2,KIF11,MAD2L1,MYBL2,SPAST, TOP2A,TTK,PRC1,PKMYT1,PTTG1,T RIP13,DLGAP5,TACC3,SMC2,SPAG5, UBE2C,ZWINT,TPX2,FBXO5,RACGA P1,NUSAP1,SPDL1,CDCA8,CEP55,ND C1,NSFL1C,KIF18B,ASPM GO:1902850 GO Biological microtubule 15 -12.89 BIRC5,CCNB1,CDC20,KIF11,MAD2L1 Processes -
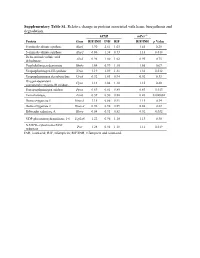
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-