DISSERTATION the Role of Rim1α in Synaptic Plasticity at the Cerebellar
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Cerebellar Microcircuit As an Adaptive Filter: Experimental and Computational Evidence
REVIEWS The cerebellar microcircuit as an adaptive filter: experimental and computational evidence Paul Dean*, John Porrill*, Carl-Fredrik Ekerot‡ and Henrik Jörntell‡ Abstract | Initial investigations of the cerebellar microcircuit inspired the Marr–Albus theoretical framework of cerebellar function. We review recent developments in the experimental understanding of cerebellar microcircuit characteristics and in the computational analysis of Marr–Albus models. We conclude that many Marr–Albus models are in effect adaptive filters, and that evidence for symmetrical long-term potentiation and long-term depression, interneuron plasticity, silent parallel fibre synapses and recurrent mossy fibre connectivity is strikingly congruent with predictions from adaptive-filter models of cerebellar function. This congruence suggests that insights from adaptive-filter theory might help to address outstanding issues of cerebellar function, including both microcircuit processing and extra-cerebellar connectivity. (BOX 2) Purkinje cell In 1967, Eccles, Ito and Szentagothai published their of spike . Simple spikes are normal action poten- 1 By far the largest neuron of the landmark book The Cerebellum as a Neuronal Machine , tials and are thought to be modulated by the many PFs cerebellum and the sole output which described for the first time the detailed microcir- that contact the PC dendritic tree. By contrast, complex of the cerebellar cortex. cuitry of an important structure in the brain. Perhaps the spikes are unique to PCs. The CF input to the PC is one Receives climbing fibre input and integrates inputs from most striking feature of the cerebellar cortical microcircuit of the most powerful synaptic junctions of the CNS, and parallel fibres and (BOX 1) is that Purkinje cells (PCs), which provide the sole complex spikes occur only when the CF fires. -

Prevalent Presence of Periodic Actin–Spectrin-Based Membrane Skeleton in a Broad Range of Neuronal Cell Types and Animal Species
Prevalent presence of periodic actin–spectrin-based membrane skeleton in a broad range of neuronal cell types and animal species Jiang Hea,b,c,1,2, Ruobo Zhoua,b,c,2, Zhuhao Wud, Monica A. Carrascoe,3, Peri T. Kurshanf,g, Jonathan E. Farleyh,i, David J. Simond, Guiping Wanga,b,c, Boran Hana,b,c, Junjie Haoa,b,c, Evan Hellera,b,c, Marc R. Freemanh,i, Kang Shenf,g, Tom Maniatise, Marc Tessier-Lavigned, and Xiaowei Zhuanga,b,c,4 aHoward Hughes Medical Institute, Harvard University, Cambridge, MA 02138; bDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138; cDepartment of Physics, Harvard University, Cambridge, MA 02138; dLaboratory of Brain Development and Repair, The Rockefeller University, New York, NY 10065; eDepartment of Biochemistry and Molecular Biophysics, Columbia University Medical Center, New York, NY 10032; fHoward Hughes Medical Institute, Stanford University, Stanford, CA 94305; gDepartment of Biology, Stanford University, Stanford, CA 94305; hHoward Hughes Medical Institute, University of Massachusetts Medical School, Worcester, MA 01605; and iDepartment of Neurobiology, University of Massachusetts Medical School, Worcester, MA 01605 Contributed by Xiaowei Zhuang, April 8, 2016 (sent for review March 25, 2016; reviewed by Bo Huang and Feng Zhang) Actin, spectrin, and associated molecules form a periodic, submem- structure, short actin filaments are organized into repetitive, brane cytoskeleton in the axons of neurons. For a better understand- ring-like structures that wrap around the circumference of the ing of this membrane-associated periodic skeleton (MPS), it is axon with a periodicity of ∼190 nm; adjacent actin rings are important to address how prevalent this structure is in different connected by spectrin tetramers, and actin short filaments in the neuronal types, different subcellular compartments, and across rings are capped by adducin (12). -

Glycine Transporters Are Differentially Expressed Among CNS Cells
The Journal of Neuroscience, May 1995, 1~75): 3952-3969 Glycine Transporters Are Differentially Expressed among CNS Cells Francisco Zafra,’ Carmen Arag&?,’ Luis Olivares,’ Nieis C. Danbolt, Cecilio GimBnez,’ and Jon Storm- Mathisen* ‘Centro de Biologia Molecular “Sever0 Ochoa,” Facultad de Ciencias, Universidad Autbnoma de Madrid, E-28049 Madrid, Spain, and *Anatomical Institute, University of Oslo, Blindern, N-0317 Oslo, Norway Glycine is the major inhibitory neurotransmitter in the spinal In addition, glycine is a coagonist with glutamate on postsyn- cord and brainstem and is also required for the activation aptic N-methyl-D-aspartate (NMDA) receptors (Johnson and of NMDA receptors. The extracellular concentration of this Ascher, 1987). neuroactive amino acid is regulated by at least two glycine The reuptake of neurotransmitter amino acids into presynaptic transporters (GLYTl and GLYTZ). To study the localization nerve endings or the neighboring fine glial processes provides a and properties of these proteins, sequence-specific antibod- way of clearing the extracellular space of neuroactive sub- ies against the cloned glycine transporters have been stances, and so constitutes an efficient mechanism by which the raised. lmmunoblots show that the 50-70 kDa band corre- synaptic action can be terminated (Kanner and Schuldiner, sponding to GLYTl is expressed at the highest concentra- 1987). Specitic high-affinity transport systems have been iden- tions in the spinal cord, brainstem, diencephalon, and retina, tified in nerve terminals and glial cells for several amino acid and, in a lesser degree, to the olfactory bulb and brain hemi- neurotransmitters, including glycine (Johnston and Iversen, spheres, whereas it is not detected in peripheral tissues. -

Acetylcholine Modulates Cerebellar Granule Cell Spiking by Regulating the Balance of Synaptic Excitation and Inhibition
2882 • The Journal of Neuroscience, April 1, 2020 • 40(14):2882–2894 Systems/Circuits Acetylcholine Modulates Cerebellar Granule Cell Spiking by Regulating the Balance of Synaptic Excitation and Inhibition X Taylor R. Fore, Benjamin N. Taylor, Nicolas Brunel, and XCourt Hull Department of Neurobiology, Duke University Medical School, Durham, North Carolina 27710 Sensorimotor integration in the cerebellum is essential for refining motor output, and the first stage of this processing occurs in the granule cell layer. Recent evidence suggests that granule cell layer synaptic integration can be contextually modified, although the circuit mechanisms that could mediate such modulation remain largely unknown. Here we investigate the role of ACh in regulating granule cell layer synaptic integration in male rats and mice of both sexes. We find that Golgi cells, interneurons that provide the sole source of inhibition to the granule cell layer, express both nicotinic and muscarinic cholinergic receptors. While acute ACh application can modestly depolarize some Golgi cells, the net effect of longer, optogenetically induced ACh release is to strongly hyperpolarize Golgi cells. Golgi cell hyperpolarization by ACh leads to a significant reduction in both tonic and evoked granule cell synaptic inhibition. ACh also reduces glutamate release from mossy fibers by acting on presynaptic muscarinic receptors. Surprisingly, despite these consistent effects on Golgi cells and mossy fibers, ACh can either increase or decrease the spike probability of granule cells as measured by noninvasive cell-attached recordings. By constructing an integrate-and-fire model of granule cell layer population activity, we find that the direction of spike rate modulation can be accounted for predominately by the initial balance of excitation and inhibition onto individual granule cells. -

A Theory of Cerebellar Function
NUMBERS 112. FEBRUARY 191 MATHEMATICAL BIOSCIENCES 25 A Theory of Cerebellar Function JAMES S. ALBUS Cybernetics and Subsystem Development Section Datu Techltiques Branch Goddard Space Flight Center Greenbelt, Maryland Communicated by Donald H. Perkel ____ ABSTRACT A comprehensive theory of cerebellar function is presented, which ties together the known anatomy and physiology of the cerebellum into a pattern -recognition data processing system. The cerebellum is postulated to be functionally and structurally equivalent to a modification of the classical Perceptron pattern -classification device. Itis suggested that the mossy fiber -+ granule cell -+ Golgi cell input network performs an expansion recoding that enhances the pattern -discrimination capacity and learning speed of the cerebellar Purkinje response cells. Parallel fiber synapses of the dendritic spines of Purkinje cells, basket cells, and stellate cells are all postulated to be specifically variable in response to climbing fiber activity. It is argued that this variability is the mechanism of pattern storage. It is demonstrated that, in order for the learning process to be stable, pattern storage must be accomplished principally by weakening synaptic weights rather than by strengthening them. 1. INTRODUCTION A great body of facts has been known for many years concerning the general organization and structure of the cerebellum. The regularity and relative simplicity ofthe cerebellar cortex have fascinated anatomists since the earliest days of systematic neuronanatomical observations. In just the past 7 or 8 years, however, the electron microscope and refined micro- neurophysiological techniques have revealed critical structural details that make possible comprehensive theories of cerebellar function. A great deal of the recent physiological data about the cerebellum come from an elegant series of experiments by Eccles and his coworkers. -

Control of Cerebellar Granule Cell Output by Sensory-Evoked Golgi Cell Inhibition
Control of cerebellar granule cell output by sensory-evoked Golgi cell inhibition Ian Duguid1,2,3, Tiago Branco1,4, Paul Chadderton5, Charlotte Arlt, Kate Powell6, and Michael Häusser3 Wolfson Institute for Biomedical Research and Department of Neuroscience, Physiology, and Pharmacology, University College London, London WC1E 6BT, United Kingdom Edited by Masao Ito, RIKEN Brain Science Institute, Wako, Japan, and approved September 1, 2015 (received for review May 25, 2015) Classical feed-forward inhibition involves an excitation–inhibition Results sequence that enhances the temporal precision of neuronal re- Sensory-Evoked Phasic and Spillover Golgi Cell Inhibition Precedes sponses by narrowing the window for synaptic integration. In Mossy Fiber Excitation in Granule Cells. Cerebellar granule cells the input layer of the cerebellum, feed-forward inhibition is thought receive direct phasic and indirect or “spillover” GABAergic in- to preserve the temporal fidelity of granule cell spikes during mossy put from Golgi cells (6, 16, 20, 21). To investigate the temporal fiber stimulation. Although this classical feed-forward inhibitory cir- dynamics of sensory-evoked inhibition in vivo, we recorded cuit has been demonstrated in vitro, the extent to which inhibition spontaneous and sensory-evoked excitatory (Vhold = −70 mV) shapes granule cell sensory responses in vivo remains unresolved. and inhibitory (Vhold = 0 mV) currents from the same granule Here we combined whole-cell patch-clamp recordings in vivo and cells in Crus II (Fig. 1 A–D). Granule cells were identified based dynamic clamp recordings in vitro to directly assess the impact of on their characteristic electrophysiological properties (Table S1), Golgi cell inhibition on sensory information transmission in the depth from the pial surface (>250 μm), and morphology (Fig. -

Resonant Synchronization in Heterogeneous Networks of Inhibitory Neurons
The Journal of Neuroscience, November 19, 2003 • 23(33):10503–10514 • 10503 Behavioral/Systems/Cognitive Resonant Synchronization in Heterogeneous Networks of Inhibitory Neurons Reinoud Maex and Erik De Schutter Laboratory of Theoretical Neurobiology, Born-Bunge Foundation, University of Antwerp, B-2610 Antwerp, Belgium Brain rhythms arise through the synchronization of neurons and their entrainment in a regular firing pattern. In this process, networks of reciprocally connected inhibitory neurons are often involved, but what mechanism determines the oscillation frequency is not com- pletely understood. Analytical studies predict that the emerging frequency band is primarily constrained by the decay rate of the unitary IPSC. We observed a new phenomenon of resonant synchronization in computer-simulated networks of inhibitory neurons in which the synaptic current has a delayed onset, reflecting finite spike propagation and synaptic transmission times. At the resonant level of network excitation, all neurons fire synchronously and rhythmically with a period approximately four times the mean delay of the onset of the inhibitory synaptic current. The amplitude and decay time constant of the synaptic current have relatively minor effects on the emerging frequency band. By varying the axonal delay of the inhibitory connections, networks with a realistic synaptic kinetics can be tuned to frequencies from 40 to Ͼ200 Hz. This resonance phenomenon arises in heterogeneous networks with, on average, as few as five connec- tions per neuron. We conclude that the delay of the synaptic current is the primary parameter controlling the oscillation frequency of inhibitory networks and propose that delay-induced synchronization is a mechanism for fast brain rhythms that depend on intact inhibitory synaptic transmission. -

GABAA,Slow: Causes and Consequences Marco Capogna1 and Robert A
Review GABAA,slow: causes and consequences Marco Capogna1 and Robert A. Pearce2 1 MRC Anatomical Neuropharmacology Unit, Mansfield Road, Oxford OX1 3TH, UK 2 Department of Anesthesiology, University of Wisconsin, Madison, WI 53711, USA GABAA receptors in the CNS mediate both fast synaptic current with a decay constant of > 30 ms. We focus pri- and tonic inhibition. Over the past decade a phasic marily on data obtained in the hippocampus because this is current with features intermediate between fast synap- the area where it has been studied most intensively and tic and tonic inhibition, termed GABAA,slow, has received where the distinction between fast and slow IPSCs ob- increasing attention. This has coincided with an served in individuals neurons is the clearest. However, it ever-growing appreciation for GABAergic cell type diver- should be noted that phasic IPSCs display a broad range of sity. Compared with classical fast synaptic inhibition, kinetics, some of which are intermediate between those of GABAA,slow is slower by an order of magnitude. In this fast and slow IPSCs [16,17]. Direct comparison between review, we summarize recent studies that have en- studies is complicated by the impact on IPSC kinetics of the hanced our understanding of GABAA,slow. These include recording conditions – including the concentration of chlo- the discovery of specialized interneuron types from ride [18], the voltage [19], and the different methods of which this current originates, the factors that could kinetic characterization and analysis [15,20–22]. Never- underlie its characteristically slow kinetics, its contribu- theless, consistent and clear distinctions between different tion to specific aspects of integrative function and net- kinetic classes of IPSCs indicate that fast and slow IPSCs work oscillations, and its potential usefulness as a novel are not simply the two ends of a single broad distribution; drug target for modulating inhibitory synaptic transmis- instead these arise from distinct classes of presynaptic sion in the CNS. -
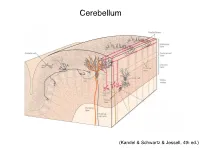
Cerebellum and Activation of the Cerebellum IO ST During Nonmotor Tasks
Cerebellum (Kandel & Schwartz & Jessell, 4th ed.) Granule186 cells are irreducibly smallChapter (6-8 7 μm) stellate cell basket cell outer synaptic layer PC rs cap PC layer grc Go inner 6 μm synaptic layer Figure 7.15 Largest cerebellar neuron occupies more than a 1,000-fold greater volume than smallest neuron. Thin section (~1 μ m) through monkey cerebellar cortex. Purkinje cell body (PC) and nucleus are far larger than those of granule cell (grc). The latter cluster to leave space for mossy fiber terminals to form glomeruli with grc dendritic claws and space for Golgi cells (Go). Note rich network of capillaries (cap). Fine, scat- tered dots are mitochondria. Courtesy of E. Mugnaini. brain ’ s most numerous neuron, this small cost grows large (see also chapter 13). Much of the inner synaptic layer is occupied by the large axon terminals of mossy fibers (figures 7.1 and 7.16). A terminal interlaces with multiple (~15) dendritic claws, each from a different but neighboring granule cell, and forms a complex knot (glomerulus ), nearly as large as a granule cell body (figures 7.1 and 7.16). The mossy fiber axon fires at an unusually high mean rate (up to 200 Hz) and is therefore among the brain’ s thickest (figure 4.6). To match the axon’ s high rate, a terminal expresses 150 active zones, 10 per postsynaptic granule cell (figure 7.16). These sites are capable of driving … and most expensive. 190 Chapter 7 10 4 8 3 9 19 10 10 × 6 × 2 2 4 ATP/s/cell ATP/s/cell ATP/s/m 1 2 0 0 astrocyte astrocyte Golgi cell Golgi cell basket cell basket cell stellate cell stellate cell granule cell granule cell mossy fiber mossy fiber Purkinje cell Purkinje Purkinje cell Purkinje Bergman glia Bergman glia climbing fiber climbing fiber Figure 7.18 Energy costs by cell type. -

Pathway Specific Drive of Cerebellar Golgi Cells Reveals Integrative Rules of Cortical Inhibition
bioRxiv preprint doi: https://doi.org/10.1101/356378; this version posted June 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Pathway specific drive of cerebellar Golgi cells reveals integrative rules of cortical inhibition Sawako Tabuchi1, Jesse I. Gilmer1,2, Karen Purba1, Abigail L. Person1 1. Department of Physiology & Biophysics 2. Neuroscience Graduate Program, University of Colorado Denver University of Colorado School of Medicine Aurora, CO 80045 Abstract: 222 words Significance Statement: 115 Introduction: 644 Discussion: 1,587 words Figures: 6 Tables: 0 Abbreviated title: Golgi cell structure-function relationship Address for Correspondence: Abigail L. Person, Ph.D. Department of Physiology & Biophysics University of Colorado School of Medicine 12800 East 19th Ave RC-1 North Campus Box 8307 Aurora, CO 80045 USA email: [email protected] (303) 724-4514 Conflict of Interest: The authors declare no competing financial interests. Acknowledgements: We thank Ms Samantha Lewis for expert technical support during the project. This work was supported by the Japan Society for The Promotion of Science (JSPS) Overseas Research Fellowship and The Uehara Memorial Foundation research fellowship to S.T.; NS084996 ; a Kingenstein Foundation fellowship; and a Boettcher foundation Webb-Waring biomedical research award to A.L.P. Imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advance Light Microscopy Core supported in part by Rocky Mountain Neurological Disorders Core Grant Number P30NS048154 and by NIH/NCATS Colorado CTSI Grant Number UL1 TR001082. Engineering support was provided by the Optogenetics and Neural Engineering Core at the University of Colorado Anschutz Medical Campus, funded in part by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number P30NS048154. -

Golgi Cell Dendrites Are Restricted by Purkinje Cell Stripe Boundaries in the Adult Mouse Cerebellar Cortex
2820 • The Journal of Neuroscience, March 12, 2008 • 28(11):2820–2826 Cellular/Molecular Golgi Cell Dendrites Are Restricted by Purkinje Cell Stripe Boundaries in the Adult Mouse Cerebellar Cortex Roy V. Sillitoe,1 Seung-Hyuk Chung,1 Jean-Marc Fritschy,2 Monica Hoy,1 and Richard Hawkes1 1Department of Cell Biology and Anatomy, Genes and Development Research Group, and Hotchkiss Brain Institute, Faculty of Medicine, The University of Calgary, Calgary, Alberta, Canada T2N 4N1, and 2Institute of Pharmacology and Toxicology, University of Zurich, CH-8057 Zurich, Switzerland Despite the general uniformity in cellular composition of the adult cerebellar cortex, there is a complex underlying pattern of parasagittal stripes of Purkinje cells with characteristic molecular phenotypes and patterns of connectivity. It is not known whether interneuron processes are restricted at stripe boundaries. To begin to address the issue, three strategies were used to explore how cerebellar Golgi cell dendrites are organized with respect to parasagittal stripes: first, double immunofluorescence staining combining anti-neurogranin to identify Golgi cell dendrites with the Purkinje cell compartmentation antigens zebrin II/aldolase C, HNK-1, and phospholipase C4; second, zebrin II immunohistochemistry combined with a rapid Golgi–Cox impregnation procedure to reveal Golgi cell dendritic arbors; third, stripe antigen expression was used on sections of a GlyT2-EGFP transgenic mouse in which reporter expression is prominent in Golgi cell dendrites. In each case, the dendritic projections of Golgi cells were studied in the vicinity of Purkinje cell stripe boundaries. The data presented here show that the dendrites of a cerebellar interneuron, the Golgi cell, respect the fundamental cerebellar stripe cytoarchitecture. -

Calcium Channel-Dependent Induction of Long-Term Synaptic Plasticity At
Research Articles: Cellular/Molecular Calcium channel-dependent induction of long- term synaptic plasticity at excitatory Golgi cell synapses of cerebellum https://doi.org/10.1523/JNEUROSCI.3013-19.2020 Cite as: J. Neurosci 2021; 10.1523/JNEUROSCI.3013-19.2020 Received: 20 December 2019 Revised: 15 December 2020 Accepted: 18 December 2020 This Early Release article has been peer-reviewed and accepted, but has not been through the composition and copyediting processes. The final version may differ slightly in style or formatting and will contain links to any extended data. Alerts: Sign up at www.jneurosci.org/alerts to receive customized email alerts when the fully formatted version of this article is published. Copyright © 2021 the authors 1 Calcium channel-dependent induction of long-term synaptic plasticity 2 at excitatory Golgi cell synapses of cerebellum 3 4 F. Locatelli1*, T. Soda1,3*, I. Montagna1, S. Tritto1, L. Botta4, F. Prestori1, E. D'Angelo1,2 5 6 1Department of Brain and Behavioral Sciences, University of Pavia, Pavia, Italy 7 2Brain Connectivity Center, IRCCS Mondino Foundation, Pavia, Italy 8 3Museo Storico della Fisica e Centro Studi e Ricerche Enrico Fermi, Rome, Italy 9 4Department of Biology and Biotechnology "L. Spallanzani", University of Pavia, Pavia, Italy 10 * FL and TS are co-first authors 11 12 Correspondence should be addressed to: 13 Prof. Egidio D'Angelo 14 [email protected] 15 and 16 Dr. Francesca Prestori 17 [email protected] 18 Dept of Brain and Behavioral Sciences 19 via Forlanini 6, 27100 Pavia, Italy 20 [email protected] 21 22 Number of pages: 25 23 Number of tables: 1 24 Number of figures: 7 25 Number of words: 209-Abstract, 546-Introduction, 1325-Discussion 26 27 Running Title: Golgi cell bidirectional plasticity 28 Key words: cerebellum, Golgi cell, synaptic plasticity, Ca2+ channels 29 1 30 Acknowledgments.