Thalassemia Minor for Severe Aplastic Anemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Severe Aplastic Anemia
Severe AlAplas tic AiAnemia Monica S. Thakar, MD Pediatric BMT Medical College of Wisconsin Outline of Talk 1. Clinical description of aplastic anemia 2. Data collection forms And a couple of quizzes in between… CLINICAL DESCRIPTIONS What is aplastic anemia? • More than just anemia – Involves low counts in 2 of 3 cell lines: red blood cells (RBC), white blood cells (WBC), pltltlatelets • Should NOT involve dysplasia – EiException: RBC can sometimes be dlidysplastic • Should have NORMAL cytogenetics • If dysplasia (beyond RBCs) or abnormal cytogenetics seen, think myelodysplastic syndrome (MDS) What is “severe” aplastic anemia? • Marrow cellularity ≤25% AND • Two of the following peripheral blood features: – Absolute neutrophil count (ANC) < 0.5 x 109/L – Platelet count <20 x 109/L – Absolute reticulocyte count < 40 x 109/L • “Very” severe aplastic anemia – Same as above except ANC <0.2 x 109/L NORMAL BttBottom Li ne: Severely Reduced HtitiHematopoietic Stem Cell Precursors SEVERE APLASTIC ANEMIA IBIn Bone M arrow Epidemiology • Half of cases seen in first 3 decades of life • Incidence: – 2 cases/m illion in WtWestern countitries – 2‐3 fold higher in Asia • Ethnic predisposition: – Asian • Genetics vs different environmental exposures? • Sex predisposition: M:F is 1:1 Pathoppyhysiology : 3 Main Mechanisms Proposed 1. Immune‐mediated – HthiHypothesis: RdRevved‐up T cells dtdestroy stem cells – Observation: Immunosuppression improves blood counts 2. Stem‐cell “depletion” or “defect” – Hypothesis: Drugs or viruses directly destroy stem cells -

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -

December Is National Aplastic Anemia Awareness Month
December Is National Aplastic Anemia Awareness Month What Is Aplastic Anemia? Aplastic anemia is a non-cancerous disease that occurs when the bone marrow stops making enough blood cells. The body makes three types of blood cells: red blood cells, which contain hemoglobin and deliver oxygen to all parts of the body white blood cells, which help fight infection platelets, which help blood clot when you bleed These blood cells are made in the bone marrow, which is the soft, sponge-like material found inside bones. The bone marrow contains immature cells called stem cells that produce blood cells. Stem cells grow into red cells, white cells, and platelets or they can make more stem cells. In patients who have aplastic anemia, there are not enough stem cells in the bone marrow to make enough blood cells. Picture of blood cells maturing from stem cells. Experts believe that aplastic anemia is an autoimmune disorder. This means that the patient’s immune system (which helps fight infection) reacts against the bone marrow and the bone marrow is not able to make blood cells. Stem cells are no longer being replaced and the left over stem cells are not working well. Therefore, the amount of red cells, white cells, and platelets begin to drop. If blood levels drop too low, a person can feel very tired (from low red cells), have bleeding or bruising (from low platelets), and/or have many or severe infections (from low white cells). What Are the Key Statistics About Aplastic Anemia? Aplastic anemia can occur in anyone of any age, race, or gender. -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -

Methemoglobinemia and Ascorbate Deficiency in Hemoglobin E Β Thalassemia: Metabolic and Clinical Implications
From www.bloodjournal.org by guest on April 2, 2015. For personal use only. Plenary paper Methemoglobinemia and ascorbate deficiency in hemoglobin E  thalassemia: metabolic and clinical implications Angela Allen,1,2 Christopher Fisher,1 Anuja Premawardhena,3 Dayananda Bandara,4 Ashok Perera,4 Stephen Allen,2 Timothy St Pierre,5 Nancy Olivieri,6 and David Weatherall1 1MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom; 2College of Medicine, Swansea University, Swansea, United Kingdom; 3University of Kelaniya, Colombo, Sri Lanka; 4National Thalassaemia Centre, District Hospital, Kurunegala, Sri Lanka; 5School of Physics, University of Western Australia, Crawley, Australia; and 6Hemoglobinopathy Research, University Health Network, Toronto, ON During investigations of the phenotypic man hypoxia induction factor pathway is There was, in addition, a highly signifi- diversity of hemoglobin (Hb) E  thalasse- not totally dependent on ascorbate lev- cant correlation between methemoglobin mia, a patient was encountered with per- els. A follow-up study of 45 patients with levels, splenectomy, and factors that sistently high levels of methemoglobin HbE  thalassemia showed that methemo- modify the degree of globin-chain imbal- associated with a left-shift in the oxygen globin levels were significantly increased ance. Because methemoglobin levels are dissociation curve, profound ascorbate and that there was also a significant re- modified by several mechanisms and may deficiency, and clinical features of scurvy; duction in plasma ascorbate levels. Hap- play a role in both adaptation to anemia these abnormalities were corrected by toglobin levels were significantly re- and vascular damage, there is a strong treatment with vitamin C. -

Aplastic Anemia Pre-HSCT Data (Form 2028)
Instructions for Aplastic Anemia Pre-HSCT Data (Form 2028) This section of the CIBMTR Forms Instruction Manual is intended to be a resource for completing the Aplastic Anemia Pre-HSCT Data Form. E-mail comments regarding the content of the CIBMTR Forms Instruction Manual to: [email protected]. Comments will be considered for future manual updates and revisions. For questions that require an immediate response, please contact your transplant center’s CIBMTR liaison. TABLE OF CONTENTS Key Fields ............................................................................................................. 2 Disease Assessment at Diagnosis ........................................................................ 2 Laboratory Studies at Diagnosis ........................................................................... 5 Transfusion Status from Diagnosis to the Start of the Preparative Regimen ........ 7 Laboratory Findings Prior to the Start of the Preparative Regimen ....................... 8 Aplastic Anemia Pre-HSCT Data Aplastic Anemia is a disease in which the bone marrow does not produce enough red blood cells, white blood cells, or platelets for the body. The disease can be idiopathic, or can be caused by environmental exposure, pharmaceutical or drug exposure, or exposure to viral hepatitis. Symptoms of aplastic anemia include, but are not limited to pallor, weakness, frequent infection, and/or easy bruising. The Aplastic Anemia Pre-HSCT Data Form is one of the Comprehensive Report Forms. This form captures aplastic -

Rituximab: Is It Possible to Link Both Autoimmune Haemolytic Anemia and Aplastic Anemia Regarding the Etiology and Management? Mina T
icine: O ed pe M n l A a r c e c e n s e s G General Medicine: Open Access Kellani MT, Gen Med (Los Angeles) 2016, 4:3 DOI: 10.4172/2327-5146.1000e110 ISSN: 2327-5146 Editorial Open access Rituximab: Is it Possible to Link Both Autoimmune Haemolytic Anemia and Aplastic Anemia Regarding the Etiology and Management? Mina T. Kelleni* Pharmacology Department, Faculty of Medicine, Minia University, Minia, Egypt *Corresponding author: Kelleni MT, Ph, M, Pharmacology department, aculty of Medicine, Minia niversity, Minia, Egypt, Tel: 201200382422; E- mail: [email protected] Rec Date: June 28, 2016; Acc Date: June 29, 2016; Pub Date: June 30, 2016 Copyright: © 2016 Kelleni MT. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Citation: Mina Kellani T (2016) Rituximab: Is it Possible to Link Both Autoimmune Haemolytic Anemia and Aplastic Anemia Regarding the Etiology and Management? Gen Med (Los Angeles) 4: e110. Editorial References In the past few years, rituximab has been recognized as a safe and 1. Abadie K, Hege KM (2014) Severe refractory autoimmune hemolytic effective emerging treatment for autoimmune hemolytic anemia anemia with five-year complete hematologic response to third course of (AIHA) [1,2]. Rituximab was also considered as a preferred second- treatment with rituximab: a case report. Journal of medical case reports 8: line therapy of warm antibody hemolytic anemia in adults in some 175. major European centers and it was shown that second-line treatment 2. -

AAMDSIF Aplastic Anemia Presentation
Aplastic Anemia Emma Groarke, MD, FRCPath Staff Clinician, Bone Marrow Failure Service National Heart, Lung, and Blood Institute National Institutes of Health AA MDS 18th May, 2019 Introduction . Aplastic Anemia . Immune mediated . What is it and what causes it? . Diagnosis . Treatment . Immunosuppression . Transplant . Pure Red Cell Aplasia . What is it and what causes it? . Ongoing research 2 A historical disease Aplastic Anemia . Rare blood disorder . 2-3 per million / year . Low blood counts and empty bone marrow . Peak age distributions . 10-25 years old and >60 years old 4 Causes of Aplastic Anemia . Immune system destroying bone marrow cells . “idiopathic” . 70% . Inherited abnormal genes . Telomere diseases . Fanconi anemia . Bone marrow toxins . Rare Young, N. S. (2018). "Aplastic Anemia." N Engl J Med 379(17): 1643-1656. 5 Mechanism of Aplastic Anemia . Immune system attacks the bone marrow . Active lymphocytes (T cells) . Increase in inflammation . Cells die Young, N.S. et al. Blood.2006 6 Aplastic Anemia Young, N. S. (2018). "Aplastic Anemia." N Engl J Med 379(17): 1643-1656. Marrow is “empty” Courtesy of Dr. Stanley Schreier, American Society of Hematology Image Bank 8 How does it affect the patient? . Neutropenia (low white cells) . Risk of infection - If less than 500 – risk of infection - If less than 200 – high risk of infection . Thrombocytopenia (low platelets) . Risk of bleeding - Risk of bleeding with trauma / procedures if <50 - Some risk bleeding <20 - Higher risk bleeding <10 – Platelet transfusion . Anemia (low red cells or hemoglobin) . Red cells carry oxygen in the blood - Symptoms include: - Tiredness - Shortness of breath . If hemoglobin <7 or symptoms - blood transfusion 9 Clonal evolution . -
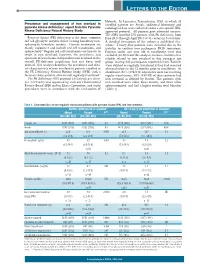
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin. -

Laboratory Features of Cutaneous Lupus Erythematosus 313
CHAPTER 23 Laboratory Features 23 of Cutaneous Lupus Erythematosus Shuntaro Shinada, Daniel J. Wallace Assuming that patients with cutaneous lupus erythematosus (CLE) do not fulfill the American College of Rheumatology criteria for systemic LE (SLE) (Tan et al. 1982), what laboratory features should the clinician look for? Interestingly, there are several. This chapter attempts to elucidate the laboratory abnormalities associated with CLE, the most common being hematologic (anemia and leukopenia), erythrocyte sedi- mentation rate (ESR), antinuclear antibodies (ANAs), and antiphospholipid anti- bodies. These features are compared with those observed in SLE and certain cuta- neous clinical subsets that have been studied. General Approach to Laboratory Testing When a dermatologist, family practitioner, internist, or rheumatologist first sees a patient with CLE, their principal concern should be to rule out evidence of systemic disease. After all, 15% of patients with CLE progress to SLE in 10–15 years of obser- vation (Rowell 1984). In addition to a complete medical history and physical exami- nation, clinical laboratory findings can be very helpful in this regard. Specifically, a blood chemistry panel allows screening for renal or hepatic involvement. Creatine phosphokinase testing assists in ruling out muscle inflammation. Evidence for autoimmune hemolytic anemia or thrombocytopenia is looked for in the complete blood cell count as well as in the lactic dehydrogenase, reticulocyte count, Coombs’ direct antibody testing, serum haptoglobin, and antiplatelet antibodies. A routine urinalysis free of cellular casts or protein makes it highly unlikely that the kidney is involved. Specific autoantibodies, almost never observed in CLE, if found, can suggest central nervous system disease (antiribosomal P,antineuronal), mixed connective tis- sue disease (anti-RNP), or other disease subsets. -

Not So Benign Hematology: Aplastic Anemia, PNH And
Not So Benign Hematology Robert A. Brodsky, MD Johns Hopkins Family Professor Director of Adult Hematology Disclosures • Dr. Brodsky serves as a Scientific Advisory Board member to: – Alexion Pharmaceuticals – Achillion Pharmaceutical – Apellis Pharmaceuticals • Grant funding: Alexion Organization • Bone Marrow Failure – Aplastic anemia • Is the era of IST coming to an end? – New complement inhibitors for PNH? • Thrombotic Microangiopathies – aHUS • Modified Ham to distinguish aHUS from TTP • Do aHUS patients need lifelong therapy with eculizumab Severe Aplastic Anemia Definitive management • Allogeneic bone marrow transplantation • Immunosuppressive therapy Acquired SAA is an Acute and Chronic Disease Take Home: PIGA Autoimmune attack on stem cells AA PNH Predisposes to clonal escape/malignancy 6pLOH 10~12% DNMT3A ASLX1 Monosomy 7 MDS Severe Aplastic Anemia • First line therapy – BMT • Matched sibling donor • Matched unrelated donor – IST (ATG/CSA) • Refractory Disease (poor response/prognosis) – Eltrombopag – Alternative donor BMT – Androgens – Other IST ATG/CSA: Late complications Rosenfeld et. al, Jama 2003;289: 1130-35 Risk of relapse > 40% in Risk of clonal evolution responders 30% failure-free survival Refractory SAA 2yr mortality approaches 50% • Repeat IST – low response rate • Eltrombopag • BMT – usually from a MUD or alternative donor Eltrombopag for Refractory SAA Desmond et al, Blood 2014 • 43 patients with refractory SAA – 1o endpoint: response at 3mos. in at least 1 linage – Response criteria (e.g, doubling of ANC, decrease in PRBCs) • Results: – 17/43 (40%) at 3 mos – 9/43 (21%) met standard response criteria at 6 mos – 7/43 (16%) trilineage response • Relapse: 3/17 (18%) responders within a year • MDS: 8/43 (16%) within 1 year Responses to eltrombopag by lineage. -

Aplastic Anemia: Diagnosis and Treatment Gabrielle Meyers, MD, and Curtis Lachowiez, MD
Clinical Review Aplastic Anemia: Diagnosis and Treatment Gabrielle Meyers, MD, and Curtis Lachowiez, MD year. 2,3 A recent Scandinavian study reported that the in- ABSTRACT cidence of aplastic anemia among the Swedish popula- Objective: To describe the current approach to diagnosis tion is 2.3 cases per million individuals per year, with a and treatment of aplastic anemia. median age at diagnosis of 60 years and a slight female 2 Methods: Review of the literature. predominance (52% versus 48%, respectively). This data is congruent with prior observations made in Barcelona, Results: Aplastic anemia can be acquired or associated with an inherited marrow failure syndrome (IMFS), where the incidence was 2.34 cases per million individu- and the treatment and prognosis vary dramatically als per year, albeit with a slightly higher incidence in males between these 2 etiologies. Patients may present along compared to females (2.54 versus 2.16, respectively).4 The a spectrum, ranging from being asymptomatic with incidence of aplastic anemia varies globally, with a dispro- incidental findings on peripheral blood testing to life- portionate increase in incidence seen among Asian pop- threatening neutropenic infections or bleeding. Workup ulations, with rates as high as 8.8 per million individuals and diagnosis involves investigating IMFSs and ruling per year.3-5 This variation in incidence in Asia versus other out malignant or infectious etiologies for pancytopenia. countries has not been well explained. There appears to Conclusion: Treatment outcomes are excellent with modern be a bimodal distribution, with incidence peaks seen in supportive care and the current approach to allogeneic young adults and in older adults.2,3,6 transplantation, and therefore referral to a bone marrow transplant program to evaluate for early transplantation is Pathophysiology the new standard of care for aplastic anemia.